
Abstract
A simple and convenient one-step hydrothermal process for synthesizing nitrogen-doped graphene/p-aminophenol composite by using ethylenediamine, p-aminophenol, and graphene oxide is described. The p-aminophenol does not only act as a spacer to prevent the graphene sheets from aggregating and restacking during the hydrothermal process but also enhances the contribution of pseudocapacitance, which further improves the performance of the electrode materials. The field emission scanning electron microscopy, Raman spectroscopy, X-ray photoelectron spectroscopy, X-ray diffraction, and electrochemical workstation are used to characterize the materials. The as-produced composite material shows superior specific capacitance of 365.7 F g−1 at a scan rate of 10 mV s−1 and excellent electrochemical cycle stability.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Owing to the excellent energy storage performance such as fast charge-discharge rates, good temperature tolerance, and safe operation, supercapacitors have attracted considerable attentions over the past decades for their potential applications in areas ranging from portable electronic products to satellites [1–5]. However, compared with secondary batteries, the relatively low energy density prevents supercapacitors’ wide applications [6]. Therefore, it is imperative to improve the energy performance in order to extend their applications in potential areas.
Currently, conventional materials mainly used as supercapacitor electrodes are carbon materials, conducting polymers and transition metal oxides [7]. Generally speaking, carbon material-based electrical double-layer capacitors have higher cycle stability, but the electrical conductivity and capacitance values are lower than metal oxides and conducting polymer-based pesudocapacitors [8–10].
Graphene, a two-dimensional (2D) system of carbon nanostructure, has attracted increasing research interests for applications in supercapacitors due to its excellent electrical conductivity, mechanical flexibility, and high theoretical surface area [11–14]. However, like other carbon materials, graphene also suffers from less satisfactory capacitance [6]. Recent studies have shown that chemical doping with heteroatoms, such as N, O, and B, can improve the electrochemical properties of graphene-based capacitors effectively [15]. Unfortunately, the individual graphene sheets tend to form irreversible aggregation or restacking during the hydrothermal reduction process because of the strong π-π interaction, resulting in a dramatic decreased surface area, reduced diffusion rate, and lower electrochemical performance [16, 17]. So far, many methods and feasible active species have been used to prevent the graphene sheets from aggregating and restacking. Yan et al. [17] fabricated highly crumpled graphene sheets (HCGSs) with high specific surface area and large pore volume through rapidly freezing a chemically reduced graphene oxide (CRG) aqueous dispersion with liquid nitrogen. But the specific capacitance is only 212 F g−1 at a scan rate of 5 mV s−1. Ai et al. [18] reported an efficient method for the synthesis of functionalized graphene materials with less aggregation. The specific capacitance is 730 F g−1 at the current density of 0.1 A g−1, but the value has a sharp decrease when the current density enhanced, which is 296 F g−1 at the current density of 0.8 A g−1. Therefore, how to minimize the restacking effectively meanwhile maintain a satisfactory electrochemical properties is of great importance.
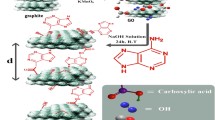
Herein, we first report a simple and convenient one-step hydrothermal process to synthesis nitrogen-doped graphene/p-aminophenol (GN/PAP) for storing energy as supercapacitor electrode using ethylenediamine, PAP, and graphene oxide (GO). The schematic illustration for the synthesis mechanism of the GN/PAP composite is shown in Fig. 1. During the hydrothermal reaction, ethylenediamine molecules continually react with GO in order to realize effective N-doping. Simultaneously, the reducible functional groups in PAP molecules also reacting with the oxygen-containing functional groups of GO. The PAP molecules are acting as spacers to control the aggregation and restacking of graphene sheets and forming loose layered structures, which facilitates hydronium ions diffusion into the layers as well as electron transport throughout the entire graphene framework. Moreover, the anchored PAP molecules greatly enhance the contribution of pseudocapacitance, which further enhanced the performance of the electrode materials.

Schematic illustration for the synthesis mechanism of the GN/PAP composite
Experiments
Preparation of GO
GO aqueous dispersion was prepared by oxidation and exfoliation of natural graphite under acidic condition by a modified Hummer’s method [19].
Synthesis of GN/PAP composite and GN
All the chemicals were analytical regent and used without further purification. Before the hydrothermal process, 50 μL of ethylenediamine and 30 mL of homogeneous GO (1 mg/mL) suspension were mixed in a flask and then put under ultrasonic agitation for 20 min; then 0.5 g PAP was added in the flask and went on ultrasonic stirring for 20 min. Finally, the mixed solution was transferred into an autoclave with a volume of 50 mL. Hydrothermal process was treated at 180 °C for 12 h. After the autoclave was naturally cooled to room temperature, washing the as-prepared black product with deionized water was done three times in order to remove residual unreacted compounds. Finally, the sample was vacuum-dried for further characterization. The GN also prepared by the above process without adding PAP.
Structure characterization
The materials were analyzed by field emission scanning electron microscopy (FESEM), transmission electron microscope (TEM), simultaneous thermal analyzer (TGA/DSC), X-ray photoelectron spectrometer (XPS), microscopic confocal laser Raman spectrometer (RAMAN), and X-ray diffractometer (XRD).
Electrochemical characterization
All electrochemical tests were carried out in a standard three-electrode cell configuration with a platinum foil as the counter electrode and a saturated calomel reference electrode (SCE). The electrolyte is 1 mol/L H2SO4 solution.
For fabricating the working electrodes, active materials, acetylene black, and polyvinylidene fluoride were mixed in a mass ratio of 80:10:10 and then dissolved in N-methyl pyrrolidone to form a slurry. Then the resulting slurry was coated onto the gauze platinum (1 × 1 cm), which was followed by drying at 40 °C for 12 h in a vacuum drying oven. The mass of active material is 6.64 and 4.16 mg for GN-based electrode and FGN-based electrode, respectively.
Results and discussion
The morphology of GN and GN/PAP composite were characterized by FESEM (the accelerating voltage is 5.0 kV) and TEM. As shown in Fig. 2a, the FESEM image of GN reveals that the vacuum-dried GN nanosheets are tightly aggregated. This aggregate structure results in a dramatic decreased surface area and reduces diffusion rate. However, the GN/PAP shows an extremely loose layered and silk-like structure (Fig. 2b), indicating PAP molecules have great contribution to the prevention of restacking of graphene sheets. This structure of GN/PAP can facilitate hydronium ions diffusion into the outer and inner areas of graphene sheets and contribute to the property of the capacitance. Actually, the surface area of GN and GN/PAP was measured by Brunauer-Emmett-Teller (BET), and the nitrogen adsorption-desorption isotherms were shown in Fig. 3a. The surface area is 201 and 481 m2 g−1, respectively, which corresponds with the FESEM results.

a FESEM image of GN reveals that the vacuum-dried GN nanosheets are tightly aggregated. b FESEM image of GN/PAP shows an extremely loose layered and silk-like structure

a Nitrogen adsorption-desorption isotherms of GN and GN/PAP. b Thermal stability of the GN and GN/PAP
The thermal stability of the GN and GN/PAP was studied by TGA/DSC (Fig. 3b). It is clearly seen that both the composites have a little mass loss around 100 °C due to the removal of residual moisture. As for GN, a steady weight loss which appeared higher than 195 °C was ascribed to the removable of oxygenated functional groups and nitrogenated functional groups. In comparison, the GN/PAP composite shows parallel weight loss curve after 300 °C indicating similar decomposition rates. However, in the temperature range from 160 to 300 °C, what appears to be a dramatic mass loss was due to the decomposing of p-aminophenol. From Fig. 3b, we can estimate that the content of p-aminophenol is 7 %.
Raman spectroscopy is a powerful technique for characterizing carbonaceous materials. The Raman spectra of GN and GN/PAP are shown in Fig. 4a at an excitation wavelength of 532 nm. The broad peak around 1345 cm−1 attributes to the D band which is associated with structural defects and partially disordered structures of the sp2 domains [20]. Similarly, the peak around 1580 cm−1 is called G band that is related to the E2g vibration mode of sp2 carbon domains which can usually be used to explain the degree of graphitization [20, 21]. The D band and G band are both the characteristic peaks of graphene-based materials. It is calculated that the intensity ratio ID/IG of GN is 1.23, but the ratio of GN/PAP is 1.17. The value was reduced, which may due to the PAP molecules anchored on the graphene basal plane. The peak located around 1190 cm−1 is associated with the C-H in-plane bending of the quinoid type ring [22]. The peak that appeared around 1282 cm−1 can be assigned to the C-N stretching vibration of the benzeniod ring, and another one centered at 1501 cm−1 is due to the C=N stretching vibration of the quinonoid ring in PAP [23]. The results of Raman spectra indicate that PAP molecules are successfully modified on the surface of GN.

a Raman spectra of GN and GN/PAP. b Full spectrum of GN/PAP. c Four main peaks from the high resolution of C1s. d Specific content of nitrogen
XPS is a significant method to research the composition of each element as well as the functional groups. As shown in Fig. 4b, the full spectrum of GN/PAP reveals the presence of C1s, N1s, and O1s; the nitrogen content exhibited in this material is 9.83 at.%. From the high resolution of C1s (Fig. 4c), four main peaks can be seen. The peaks at 284.7, 285.5, 286.2, and 288.3 eV are attributed to the sp2 carbon (C=C), C-N, C-OH, and C=O configurations, respectively [24, 25]. The specific content of nitrogen is shown in Fig. 4d. The peak at 398.7 eV is ascribed to pyridinic N, and the peak at 399.3 eV can be attributed to amine moieties or other sp3-C and nitrogen bonds. The peak at 400.2 eV can be ascribed to pyrrolic structure [15, 26, 27]. It further reveals that nitrogen element is doped into the sheets of graphene successfully. Researchers have demonstrated that nitrogen-doped active sites (pyridinic N especially) on graphene sheets can provide reaction sites for electrochemical reactions [28, 29]. These active sites have a great effect on promoting the oxidation reduction reaction of PAP molecules which are anchored on the surface of graphene sheets.
The structure of prepared materials was further characterized by X-ray diffraction (XRD) at the scanning step of 0.02626°. Figure 5 presents the XRD patterns of GO, GN, and GN/PAP composite, respectively. The XRD pattern of GO appears as a basal reflection with a strong and sharp peak at 2θ = 11.44° (d spacing = 0.773 nm). As for GN, the XRD pattern exhibits a broad peak centered at 2θ = 24.41°, corresponding to the graphitic (002) profile with an interlayer spacing of 0.365 nm. The smaller d spacing of GN than that of GO which is attributed to the oxygen-containing functional groups on the graphene sheets are partially reduced by ethylenediamine during the hydrothermal process. While the XRD pattern of GN/PAP shows a broad peak centered at 2θ = 25.32° (d spacing = 0.352 nm). The lower d value of GN/PAP than that of GN indicates p-aminophenol molecules also acting as a reducing agent for reducing the oxygen-containing functional groups of GO.

XRD patterns of GO, GN, and GN/PAP composite
Electrochemical behavior
Figure 6a shows the cyclic voltammetry (CV) curves of GN and GN/PAP composite at a scan rate of 10 mV s−1. The CV shape of the GN presents a quasi-rectangle, which indicates that GN-based materials mainly possess electrical double-layer capacitance. In contrast, the CV curve of GN/PAP composite displays a pear-like shape with two couples of distinct redox peaks, attribute to the redox transition of PAP between reduction state and oxidation state, and indicates the coexistence of both the electrical double-layer capacitance and pseudocapacitance. The specific capacitance of the electrode can be calculated according the following equation: C = (∫Idv)/(νmΔV), where C is the specific capacitance (F g−1), I means the response current (A), v is the potential (V), ΔV is the voltage drop (V), ν is the potential scan rate (mV s−1), and m represents the mass of electroactive material (g) in the electrodes. The GN-based supercapacitor shows a specific capacitance of 127.5 F g−1 at 10 mV s−1, while the GN/PAP reveals an impressive capacitance of 365.7 F g−1. Figure 6b shows the CV curves of GN/PAP composite at different scan rates of 5, 10, and 20 mV s−1, representing capacitance of 411.3, 365.7, and 316.3 F g−1, respectively.

a CV curves of GN and GN/PAP at a scan rate of 10 mV s−1. b CV curves of GN/PAP at different scan rates of 5, 10, and 20 mV s−1. c Galvanostatic charge/discharge curve of GN/PAP. d Electrochemical stability of GN/PAP at current density of 10 A g−1
The specific capacitance of the supercapacitors is also revealed by galvanostatic measurements. The galvanostatic charge/discharge curve of GN/PAP shows a deviation from the ideal triangle shape exhibited in GN (Fig. 6c). This result also confirms the inspiring contribution of pseudocapacitance. Moreover, we can obviously find that the GN/PAP-based material has a larger capacitance when compared to the charge/discharge curve of GN/PAP with GO/PAP (without adding ethylenediamine during the reaction progress). The specific capacitance of GN/PAP and GO/PAP is 474.2 and 187.5 F g−1 at the current density of 1 A g−1, respectively. This phenomenon reveals that by using N-doping, the redox reactions between the electrolyte ions and PAP molecules are reinforced. Moreover, the surface wettability and electronic conductivity have also been improved [30, 31]. Long cycle life is another parameter for supercapacitor electrode materials. The electrochemical stability of GN/PAP composite is presented in Fig. 6d at the current density of 10 A g−1. Excitingly, the capacitance retention is about 80 % after 1000 charge/discharge cycles. This phenomenon reveals that the GN/PAP-based capacitor has excellent electrochemical cycle stability.
Electrochemical impedance spectroscopy (EIS) technique is extensively used to elucidate the electrical characteristics of the electrode material and its interface with supporting electrolyte. Figure 7 shows the typical Nyquist plots of the as-prepared materials which were measured in 1 M H2SO4 solution in the frequency ranging from 0.01 Hz to 100 kHz. Based on the Nyquist plots, the best-fitting values of the equivalent circuit model shown by the inset are listed in Table 1. From the table, the series solution resistances (R S) of GN and GN/PAP are 4.518 and 4.799 Ω cm2, respectively. C 1 represents double-layer capacitance. The ionic charge-transfer resistance, R ct1, is lower for the GN/PAP-coated electrode, suggesting that this material exposes more surface area than that of GN, favorable for the exchange of electrons. This statement is further supported by the lower resistance of the Warburg element for GN/PAP (10.323 Ω cm2) in comparison with GN (22.358 Ω cm2). On the other hand, GN/PAP shows lower electron-hopping resistance (R ct2). C 2 in parallel to R ct2 represents Faradic capacitance, and it is proportional to the redox transitions in this experiment. GN/PAP-coated electrode shows that C 2 is 376.640 μF cm−2 while GN-coated one only reveals 21.749 μF cm−2. The higher C 2 attributes to the PAP molecules which were modified on the graphene sheets. Based on the above comparisons, the capacitive performances of GN/PAP are better than that of GN.

Nyquist plots of the materials
Conclusion
In summary, the GN/PAP composite was successfully synthesized via a simple and convenient one-step hydrothermal process. The loose layered and silk-like structure of GN/PAP can facilitate hydronium ions diffusion into the outer and inner areas of graphene sheets, and the anchored PAP molecules greatly enhance the contribution of pseudocapacitance. The electrochemical behavior shows that the GN/PAP composite owns outstanding electrochemical performance as an electrode for supercapacitors with an impressive capacitance of 365.7 F g−1 at a scan rate of 10 mV s−1 and excellent electrochemical stability with 92 % of its initial capacitance retained after 200 charge/discharge cycles at the current density of 10 A g−1.
References
Miller JR, Simon P (2008) Electrochemical capacitors for energy management. Science 321:651
Brownson DAC, Kampouris DK, Banks CE (2011) An overview of graphene in energy production and storage applications. J Power Sources 196:4873
Sheng KX, Sun YQ, Li C, Yuan WJ, Shi GQ (2012) Ultrahigh-rate supercapacitors based on eletrochemically reduced graphene oxide for ac line-filtering. Sci Rep 2:247
Frackowiak E, Beguin F (2001) Carbon materials for the electrochemical storage of energy in capacitors. Carbon 39:937
Gao B, Hao L, Fu QB, Su LH, Yuan CZ, Zhang XG (2010) Hydrothermal synthesis and electrochemical capacitance of RuO2·xH2O loaded on benzenesulfonic functionalized MWCNTs. Electrochim Acta 55:3681
Hall PJ, Mirzaeian M, Fletcher SI, Sillars FB, Rennie AJR, Shitta-Bey GO, Wilson G, Cruden A, Carter R (2010) Energy storage in electrochemical capacitors: designing functional materials to improve performance. Energy Environ Sci 3:1238
Lin JX, Zheng YY, Du QF, He MP, Deng ZW (2013) Synthesis and electrochemical properties of graphene/MnO2/conducting polymer ternary composite for supercapacitors. Nano 8(1):1350004-1
Stoller MD, Ruoff RS (2010) Best practice methods for determining an electrode material’s performance for ultracapacitors. Energy Environ Sci 3:1294
Sun YQ, Wu QO, Shi GQ (2011) Graphene based new energy materials. Energy Environ Sci 4:1113
Liu R, Lee SB, Am J (2008) MnO2/Poly(3,4-ethylenedioxythiophene) coaxial nanowires by one-step coelectrodeposition for electrochemical energy storage. Chem Soc 130:2942
Chen J, Sheng KX, Luo PH, Li C, Shi GQ (2012) Graphene hydrogels deposited in nickel foams for high-rate electrochemical capacitors. Adv Mater 24:4569
Zhang L, Shi GQ (2011) Preparation of highly conductive graphene hydrogels for fabricating supercapacitors with high rate capability. J Phys Chem C 115:17206
Wang L, Wang DL, Zhu JS, Liang XS (2013) Preparation of Co3O4 nanoplate/graphene sheet composites and their synergistic electrochemical performance. Ionics 19:215
Liu TT, Shao GJ, Ji MT, Ma ZP (2014) Composites of olive-like manganese oxalate on graphene sheets for supercapacitor electrodes. Ionics 20:145
Wang DW, Min YG, Yu YH, Peng B (2014) A general approach for fabrication of nitrogen-doped graphene sheets and its application in supercapacitors. J Colloid Interface Sci 417:270
Wang Y, Shi ZQ, Huang Y, Ma YF, Wang CY, Chen MM, Chen YS (2009) Supercapacitor devices based on graphene materials. J Phys Chem C 113:13103
Yan J, Xiao Y, Ning GQ, Wei T, Fan ZJ (2013) Facile and rapid synthesis of highly crumpled graphene sheets as high-performance electrodes for supercapacitors. RSC Adv 3:2566
Ai W, Zhou WW, Du ZZ, Du YP, Zhang H, Jia XT, Xie LH, Yi MD, Yu T, Huang W (2012) Benzoxazole and benzimidazole heterocycle-grafted graphene for high-performance supercapacitor electrodes. J Mater Chem 22:23439
Hummers WS, Offeman RE (1958) Preparation of graphitic oxide. J Am Chem Soc 80:1339
Kudin KN, Ozbas B, Schniepp HC, Prud’homme RK, Aksay IA (2008) Raman spectra of graphite oxide and functionalized graphene sheets. Nano Lett 8:36
Sheng ZH, Shao L, Chen JJ, Bao WJ, Wang FB, Xia XH (2011) Catalyst-free synthesis of nitrogen-doped graphene via thermal annealing graphite oxide with melamine and its excellent electrocatalysis. ASC Nano 5(6):4350
Shah AA, Holze R (2008) Spectroelectrochemistry of two-layered composites of polyaniline and poly(o-aminophenol). Electrochim Acta 53:4642
Tagowska M, Palys B, Jackowska K (2004) Polyaniline nanotubules—anion effect on conformation and oxidation state of polyaniline studied by Raman spectroscopy. Synth Met 142:223
Singh D, Joung D, Zhai L, Das S, Khondaker S, Seal S (2011) Graphene based materials: past, present and future. Prog Mater Sci 56:1178
Yan J, Wei T, Shao B et al (2010) Preparation of a graphene nanosheet/polyaniline composite with high specific capacitance. Carbon 48:487
Lee JW, Ko JM, Kim JD (2012) Hydrothermal preparation of nitrogen-doped graphene sheets via hexamethylenetetramine for application as supercapacitor electrodes. Electrochim Acta 85:459
Chen P, Yang JJ, Li SS, Wang Z, Xiao TY, Qian YH, Yu SH (2013) Hydrothermal synthesis of macroscopic nitrogen-doped graphene hydrogels for ultrafast supercapacitor. Nano Energy 2:249
Qu LT, Liu Y, Baek JB, Dai LM (2010) Nitrogen-doped graphene as efficient metal-free electrocatalyst for oxygen reduction in fuel cells. ASC Nano 4:1321
Guo HL, Peng S, Xu JH, Zhao YQ, Kang XF (2014) Highly stable pyridinic nitrogen doped graphene modified electrode in simultaneous determination of hydroquinone and catechol. Sensors Actuators B Chem 193:623
Wang HB, Maiyalagan T, Wang X (2012) Review on recent progress in nitrogen-doped graphene: synthesis, characterization, and its potential applications. ACS Catal 2:781
Han J, Zhang LL, Lee S, Oh J, Lee KS, Potts JR, Ji JY, Zhao X, Ruoff RS, Park S (2013) Generation of B-doped graphene nanoplatelets using a solution process and their supercapacitor applications. ASC Nano 7:19
Acknowledgments
This study was supported by the Opening Project of CAS Key Laboratory of Materials for Energy Conversion.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Chen, C., Fan, W., Zhang, Q. et al. High performance of supercapacitor based on nitrogen-doped graphene/p-aminophenol electrodes. Ionics 21, 2639–2645 (2015). https://doi.org/10.1007/s11581-015-1456-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-015-1456-5