
Abstract
Physiologically-based pharmacokinetic (PBPK) modeling is a popular drug development tool that integrates physiology, drug physicochemical properties, preclinical data, and clinical information to predict drug systemic disposition. Since PBPK models seek to capture complex physiology, parameter uncertainty and variability is a prevailing challenge: there are often more compartments (e.g., organs, each with drug flux and retention mechanisms, and associated model parameters) than can be simultaneously measured. To improve the fidelity of PBPK modeling, one approach is to search and optimize within the high-dimensional model parameter space, based on experimental time-series measurements of drug distributions. Here, we employ Latin Hypercube Sampling (LHS) on a PBPK model of PEG-liposomes (PL) that tracks biodistribution in an 8-compartment mouse circulatory system, in the presence (APA+) or absence (naïve) of anti-PEG antibodies (APA). Near-continuous experimental measurements of PL concentration during the first hour post-injection from the liver, spleen, kidney, muscle, lung, and blood plasma, based on PET/CT imaging in live mice, are used as truth sets with LHS to infer optimal parameter ranges for the full PBPK model. The data and model quantify that PL retention in the liver is the primary differentiator of biodistribution patterns in naïve versus APA+ mice, and spleen the secondary differentiator. Retention of PEGylated nanomedicines is substantially amplified in APA+ mice, likely due to PL-bound APA engaging specific receptors in the liver and spleen that bind antibody Fc domains. Our work illustrates how applying LHS to PBPK models can further mechanistic understanding of the biodistribution and antibody-mediated clearance of specific drugs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Drug development generally involves extensive studies in animal models to assess the pharmacokinetics and biodistribution of the drug over time, as the complex interactions between the drug and different elements of a living system must be carefully assessed, and they cannot be properly assessed by in vitro experiments alone. To interpret and guide animal studies, physiologically-based pharmacokinetic (PBPK) models are increasingly used to integrate drug and system (physiology) information into a mathematical modeling framework to describe and predict the absorption, distribution, metabolism, and excretion of the drug in humans and animals. The PBPK models are further integrated with data from animal studies. A key feature of PBPK models is their mechanistic focus, capturing the mass balance of the drug of interest by incorporating the anatomical, physiological, physical, and chemical processes that govern the fate of a drug in the body over time. Many PBPK parameters are pre-determined experimentally—for example, organ-specific blood flow rates and volumes have been determined using appropriate radioactive tracers for a variety of model organisms (Brown et al. 1997; Davies and Morris 1993; Garg and Balthasar 2007; Kaliss and Pressman 1950). PBPK models, coupled with the principle of allometric scaling, enable translations across species and populations (Huh et al. 2011; Mordenti et al. 1991; Ritschel et al. 1992). Not surprisingly, PBPK models are broadly adopted in drug development and regulation (Jones and Rowland-Yeo 2013; Yuan et al. 2019), including the prediction of drug-drug interactions (Stader et al. 2021; Ueno et al. 2021), dose adjustment in special populations (Lang et al. 2020; Lutz et al. 2021), bioequivalence assessment of complex drug formulations (Fan et al. 2017; Le Merdy et al. 2020), and many other mechanistic explorations (Cao and Jusko 2012; He et al. 2018, 2019; Nasu et al. 2005; Walsh et al. 2016).
A limiting aspect of PBPK models is that some, and often many, key parameters are either not accurately measured or not well-documented. This is especially true in the development of new therapeutics. The complexity of living systems, as reflected by a large number of model parameters together with ethical as well as practical limitations, makes it difficult to measure or estimate all parameter values. This necessitates the use of various mathematical and statistical techniques: to identify the most sensitive and influential model parameters that govern drug efficacy and uptake to the target; and, to estimate values and ranges of model parameters to improve the predictive power of the PBPK model. Examples include assessing variable response to treatment and individual-to-individual variability (Edginton et al. 2016; Krauss et al. 2013; Tsamandouras et al. 2015).
We previously developed a minimal, 2-compartment PBPK model to capture the systemic circulation over time of liposomal drug carriers modified with polyethylene glycol (PEG), or PEGylated liposomes (PL), in the presence of anti-PEG antibodies (APA) (McSweeney et al. 2018). Clinically, APA have been shown to bind to a number of PEGylated drugs. Above a critical threshold, APA cause rapid elimination of the PEGylated drugs from the circulation, greatly reducing their efficacy as well as increasing the adverse events associated with the therapy (Abu Lila et al. 2013; Yang and Lai 2015). We expanded the previous minimal PBPK model to 8 compartments in order to improve the resolution of the biodistribution of PEG-liposomes, with and without APA. We compare the 8-component PBPK model predictions to experimental data obtained from tracing radiolabeled PL in different organ tissues over time, using positron emission tomography/computed tomography (PET/CT) scanning. Here, using the experimental time series data as truth sets, we employ Latin Hypercube Sampling (LHS) on the 8-compartment PBPK model for both APA+ and naïve mice. This allows us to infer model parameter ranges, identify the primary parameters that induce the largest model variability, and optimize parameter values that give best fits to the experimental truth set. In doing so, we confirm hypotheses that the liver and spleen are most responsible for accelerated blood clearance (ABC) of PEGylated liposomes by APA. Further, we identify the specific kinetic mechanisms, drug retention by the liver and spleen that is amplified by the presence of APA, as the driving factors for increased clearance.
2 Model Parametrization and Exploration
2.1 Parametrizing the Model with Latin Hypercube Sampling and Experimental Data
Here we introduce a full, multi-compartment PBPK model to describe the biodistribution of PL, and how it is altered in the presence of APA. The model was able to fit mouse data with and without APA, illustrating its capacity to recapitulate the accelerated blood clearance (ABC) phenomenon mediated by APA. All mice in this study exhibited high APA titers (> 15 µg/ml anti-PEG IgG) induced by injection of empty PEG-liposomes at least one week prior to the study. This represents APA well above the threshold needed for ABC (McSweeney et al. 2018). The model accounts for the redistribution of radiolabeled PL from an IV injection through the lungs, liver, spleen, kidney, and muscle. A remainder compartment comprises the brain, adipose, and other tissues that may sequester small amounts of the drug. Initializing the model at t = 1 min allows us to model the complete redistribution of drug to the appropriate organs, with some degree of noise, while eliminating excessive fluctuations due to very fast transient changes immediately post injection and limitations in the timing of the PET/CT imaging used to obtain the experimental data (i.e., a peak in signal that may appear and disappear between the 10-s windows used for binning images).
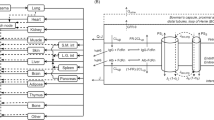
We consider the PBPK system (Fig. 1, Eqs. 1–8) defined by the following system of rate equations for transport of the “Drug” (PL) between all compartments (p = venous plasma, li = liver, k = kidney, s = spleen, m = muscle, a = arterial plasma, lu = lung, r = remainder compartment):

Schematic of the PBPK model. The PBPK system tracks the concentration of PL in each organ compartment after an initial IV injection
Here Qx represents blood flow in compartment x, Vx represents the volume of tissue and interstitial components, Cx represents the concentration of PL (in %ID/g), CLx represents clearance rate, frx represents the dimensionless permeability fraction controlling the extravasation rate into compartment x, and Kpx represents the partitioning (drug retention) coefficient and the potential for accumulation in compartment x. A model incorporating permeability-limited distribution was chosen because the PL are ~ 100 nm in diameter, a sufficiently large size to make its extravasation rate much lower than blood perfusion. The concentration in each compartment was taken as the weighted average of the tissue/ interstitial sub-compartment and the plasma sub-compartment, assuming that each sub-compartment was well-mixed, which corresponded with the overall PET signal in each region.
In order to explore ranges for all parameters, we used Latin Hypercube Sampling (LHS). Rather than running parameter sweeps across the product space of ranges for all parameters, which is computationally impractical, LHS semi-randomly samples the product space of parameter ranges (McKay et al. 1979; Iman et al. 1981). We parametrized the system at the individual mouse level in order to consider inter-mouse and inter-cohort variability (Helton and Davis 2003; Hora and Iman 1989; Marino et al. 2008), and progressively refined parameter ranges based on LHS outputs for each in silico mouse.
To set limits of minimum and maximum possible parameter values, we first searched the literature. Some physiological parameters are well-documented in the literature, for example, blood flow and organ volume (Brown et al. 1997; Davies and Morris 1993; Garg and Balthasar 2007; Kaliss and Pressman 1950). Further details such as residual blood volume allowed us to determine an even more accurate approximation of the signal or drug concentration in each compartment (Brown et al. 1997; Kaliss and Pressman 1950). We selected a suitable initial range for the drug retention coefficients (Kpx) per compartment from data, as the nearly continuous PET scan provided sufficient data to estimate Kpx for each compartment as a function of its area under the curve (AUC). AUC for an organ compartment is computed as the integral of drug concentration over the duration of the study and is used as a measure of the organ’s total drug exposure for this duration. Restrictions on values of these more well-documented parameters allowed us to perform a targeted exploration of less-known parameters, e.g., the organ permeability fraction (frx).
For the parameters with less certainty, tests were used to determine parameter bounds that result in valid vs. invalid output. These pass/fail tests progressively narrowed the parameter ranges in order to more reliably produce acceptable outputs. For the first round of tests, a parameter set was considered successful (passed) if (1) the model drug concentration in each organ was within fivefold of in vivo experimental concentrations, (2) the AUC was within 20% of the AUC calculated from in vivo data, and (3) the remainder compartment was non-negative and thus preserved mass balance. After completing a round of LHS simulations using relatively wide ranges for the least well-known parameters, each parameter range was examined to see which values yielded simulations that passed all tests and which values yielded simulations that failed at least one test. We found a representative parameter set whose model concentration and AUC plots visually resembled the in vivo experimental data and whose parameter values were not relatively close to values found in a failed test. From this representative parameter set, we created an updated range for each parameter by extending 10% above and below its value. The sampling ranges of the refined drug-retention coefficients (Kpx) were reasonably close to the initial guess from AUC ratios (most well within \(\pm \hspace{0.17em}50\%\)). Using these updated parameter ranges, we performed a second round of LHS simulations (Figs. S.1, S.2). We sorted the model outputs (predicted drug concentration in each organ for the duration of the study) by the total sum of squared errors compared to the in vivo experimental data, and a characteristic parameter set for each mouse was selected from the 10 simulations with the lowest least-squares error. The least-squares error function was defined such that greater weight was placed on compartments with greater drug uptake, because errors were normalized by total signal in the mouse rather than normalized by individual organ. This optimization procedure prioritized organs with higher drug concentration and overall signal, which also enabled the model to converge faster and more consistently when implemented. After the parameter sets had been sorted according to the lowest least-squares error, the top 10 predictions based on LHS-randomized parameter selection tracked with the data (Fig. 2). The optimal parameter set for each mouse was selected from these candidate sets.

In vivo experiment and PBPK model comparisons. Data (black dots) show PL concentrations in 6 compartments (blood plasma and 5 primary organs) of one APA+ mouse, obtained via continuous PET/CT scan for the first hour after PL injection. Simulated data using the 10 most suitable parameter sets within the optimal ranges identified by LHS are overlaid with the experimental measurements (colored curves, with each color representing a unique LHS simulation ID) (Color figure online)
2.2 Parametrizing the Model: An Exploration of Parameter Space
Since prolonged drug circulation typically correlates with improved efficacy, we focused on the altered clearance of PL between these cohorts. We next sought to specifically characterize which process(es) and compartment(s) in the PBPK model (and corresponding physiological processes) can distinguish drug clearance to the highest degree between cohorts of mice with and without APA.
Evidence suggests that the systemic clearance of PL and small immune complexes, such as PL bound to APA, is predominantly liver-mediated (Ganesan et al. 2012; Ishida et al. 2005; McSweeney et al. 2018), in good agreement with our experimental (Fig. S.3) and model findings that the organ with greatest PL accumulation over time is the liver. To explore this in greater detail, we focused simulations on the parameters responsible for controlling the extent of drug exposure and retention in the liver: the liver permeability fraction, frli, and the retention coefficient, Kpli. Together, the parameters frli and Kpli control total drug in the liver. We used the model to explore how APA alter the role of frli and Kpli, contributing mechanistically to the increased liver accumulation.
We considered the concentration of PL in the liver as a measurement of percent injected dose per gram liver tissue (%ID/g). We measured uptake one hour post-injection, consistent with the early-phase redistribution of the liposomes and the available data for continuous PET/CT scanning. We conducted a parameter sweep for liver-specific fr and Kp, through the widest region searched with LHS, while fixing the other parameters at the average of their optimized values (generating the prediction with the lowest least-squares error) for representative naïve and APA+ mice according to the characteristic LHS runs, and thus generated a space of simulated liver uptake in an “average mouse” for each cohort 1 h after liposome injection (Fig. 3). Here, we visualize LHS-identified parameter ranges for liver permeability (frli) and retention (Kpli), and the LHS-identified best-fit values of both liver parameters from the experimental data over 1 h for naive and APA+ mice. In this state diagram, we observe regions of parameter sensitivity and impact. Assuming a base level of drug availability (frli), there is consistently a gradient of PL taken up by the liver, increasing while Kpli is increased. However, distinct regions emerge as frli is varied from its lower to upper bounds, occurring at the ridge where contour lines switch from horizontal to vertical (Fig. 3). This implies that given sufficient permeability of the drug in the liver (through frli above a sufficiently high threshold), the final liver PL concentration loses sensitivity to frli, and liver drug concentration is driven primarily by the organ’s ability to retain the PL (determined by Kpli). At the far left of the landscape, for low Kpli, liver retention is sufficiently low such that permeability (frli) makes no difference in overall uptake, since very little drug is retained regardless of availability.

PBPK-generated heat map of drug concentration in liver at 1 h post injection versus liver permeability (frli) and liver retention (Kpli) parameter specifications. Ranges of parameters are identified from Latin Hypercube Sampling (LHS) based on experimental measurements for 1 h post PEGylated liposome injection, from representative a naïve and b APA+ mice. All other PBPK model parameters are set at the average of optimal LHS-identified values for each cohort. The black dots are mean values of both liver parameters optimized by LHS based on best-fits to the experimental data over the entire 1-h measurements, whereas the ellipses have semi-axes given by the standard deviations of each LHS-identified parameter for each cohort. Heat map colors and values are normalized so that 1 = 100%ID/g (Color figure online)
When the model was optimized for the experimental data, all mice showed sufficient frli above the minimal threshold, and all naïve mice clustered on the far left of the landscape. Thus, both naïve and APA+ mice fall in the regions where Kpli is a more sensitive parameter than frli in governing liver uptake. In other words, uptake in the liver compartment is driven more by its retention ability (partitioning coefficient Kpli), and loses dependence on permeability after a certain point. In this framework, permeability simply controls how quickly the uptake occurs, but not total accumulation. For example, if Kpli is fixed at a high value (assume, for instance, Kpli > 3), frli close to 1 will result in the drug uptake reaching the organ’s “carrying capacity” (determined by Kpli) almost immediately, within the first few minutes, whereas frli < 0.1 will result in the drug concentration gradually reaching its upper limit over the course of an hour. The liver retention coefficient (Kpli) has similarly been identified as a critical parameter in the context of PBPK models for other therapeutics (Hsieh et al. 2018). Correspondingly, we also find that if the retention ability and capacity to take up a drug are high enough, the availability of the PL determined by frli then becomes the factor limiting uptake, and we see a rapidly increasing gradient as frli increases. Ultimately, if Kpli and frli are sufficiently high (in the top right corner in Fig. 3), we reach the upper bound of drug uptake in the liver. This “plateau” is limited only by the amount of drug in the body.
Physiologically, these findings suggest that a heightened drug retention in the liver (likely due to APA-mediated uptake into cells in the liver) is more responsible for increased concentration of PL in the liver in mice with APA, rather than drug permeability of the liver tissue or any changes to permeability promoting extravasation in the APA+ cohort. This is not surprising, as hepatic clearance is consistent with the elimination of small immune complexes from the circulation by liver sinusoidal endothelial cells expressing the receptor FcγIIb, which binds to the Fc domain of the PL-bound APA.
We next investigated other methods to confirm the liver drug retention capability Kpli as the key parameter in differentiating the biodistribution patterns of PL in mice with distinct levels of APA. We examined the full parameter space and found that the parameters cluster for the mice within each cohort, with small fluctuations. Furthermore, in all drug retention (Kpx) and permeability (frx) parameters, except for drug retention in the liver and spleen, there is no notable cohort-to-cohort difference as opposed to variation between individuals within a cohort; i.e., variability due to liver and spleen retention (Kpli, Kps) is only notable in the presence of APA. This suggests APA+ and naïve mice otherwise appear physiologically similar (Fig. 4, Fig. S.4, Table 1) (figure format adapted from Moses 2021). Organ-specific Kp (drug retention ability) parameters optimized by LHS cluster consistently at small values (< 0.5) with the exception of liver- and spleen-specific Kp in APA + mice. Kpli and Kps are the most variable parameters, and the variability increases with the presence of APA (Fig. 4, Fig. S.4). The naïve mice are highly robust (Fig. 4A). Organ-specific fr (extent of permeability) parameters are consistently optimized around 0.1 in all mice, regardless of APA status. Thus, drug permeability (fr) parameters have low variability relative to drug retention (Kp) parameters and are not influenced by the presence of APA (Fig. 4).

Spider plots of LHS-optimized parameters. Retention and permeability parameters for all 5 primary organs from a 6 naïve mice and b 6 APA+ mice. Each colored line represents an individual mouse (diamonds and dashed lines indicate mice with APA, and circles and solid lines indicate naïve mice). Note that spleen data are not available for 4 of the mice (Color figure online)
While spleen data were unavailable for 4 of the mice in the sample set due to inability to accurately delineate the spleen in PET/CT imaging, gamma counter readings at the terminal time point confirm a high splenic concentration. Isolating and looking more closely at the retention coefficients for liver and spleen, which reflect the net uptake of APA/nanoparticle complexes by different cells within these organs, we can identify a significant disparity in the values of LHS-optimized parameters for each mouse depending on whether or not a mouse has high levels of APA (Fig. 5). Specifically, the optimal liver and spleen retention parameters in APA+ mice were both significantly larger (p < 0.05: unpaired, one-tailed t test with Welch’s correction), which reflects markedly greater rates of net uptake by cells in these organs, and more variable (p < 0.05: F test). This is consistent with both liver (primary) and spleen (secondary) retention as the key organs responsible for the APA-induced ABC of PL. One possible physiological mechanism for such APA-induced retention of PL in liver and spleen is the direct uptake of PL/APA complexes, formed in the blood, by liver sinusoidal endothelial cells (LSECs) and Kupffer cells via FcγRIIb (Pyzik et al. 2019).

Correlations between liver and spleen LHS-optimized retention parameters. Values corresponding to APA + mice (n = 6 in liver, n = 4 in spleen) are presented as squares, and values corresponding to naïve mice (n = 6 in liver, n = 4 in spleen) are presented as circles. Error bars represent standard deviations. (*p < 0.05, ***p < 0.001)
3 Discussion
PEG is routinely conjugated to proteins and liposomes to reduce the immunogenicity and extend the systemic circulation times of the underlying therapeutic (Yang and Lai 2015). There are currently at least 12 PEGylated therapeutics on the market, and many more in development (Yang and Lai 2015). Unfortunately, evidence has emerged that some patients may possess pre-existing or develop APA, which in turn leads to ABC of select PEGylated therapeutics (Povsic et al. 2016; Kozma et al. 2020), including Krystexxa (Hershfield et al. 2014; Lipsky et al. 2014) and Oncospar (Armstrong et al. 2007). Animal studies have repeatedly shown that APA can lead to ABC of PL (Ishida et al. 2005; Mohamed et al. 2019; McSweeney et al. 2018). There are even suspicions that APA may be responsible for the anaphylactic response to COVID-19 mRNA vaccines formulated with PEG-lipids (Kleine-Tebbe et al. 2021; Worm et al. 2021). Thus, there is immense interest in a better understanding of how APA can engage PEGylated drugs in living systems, and the consequent temporal fate of the PEGylated drug. While in vivo studies have elucidated the immunological mechanisms of the APA induction (Ishida et al. 2005), the ABC phenomenon (Ishida and Kiwada 2008; Abu Lila et al. 2013), and the resulting biodistribution of the PEGylated drugs, these existing studies do not readily reveal the key physiological mechanisms that are responsible for the clearance itself.
Here, by performing search and optimization using Latin Hypercube Sampling on an 8-compartment PBPK model together with PET/CT data, we identify the most influential factors that drive the observed accumulation of PL/APA complexes in the liver and spleen. Our work gives insight into the physiological factors underlying the PBPK model for PEGylated nanomedicine biodistribution, refines the parameter unknowns from experimental data, and explains the observed variation in the system. In this way, we gain mechanistic insights into accelerated drug clearance by APA that are otherwise difficult to measure and validate experimentally.
In the model, consistent with PET/CT studies, we observed that naïve mice exhibited less variability in their optimized parametrizations and maintained nearly constant signals through the first hour. This contrasts with the greater variations in mice with appreciable APA titers, which underscores the complexity of APA-mediated ABC. The liver and spleen ability to retain the PL drug carriers (high retention coefficients Kpli, Kps) exhibited the most significant difference between the naïve and APA+ cohorts and subsequently appeared to be the driving parameters in this system, distinguishing the cohorts of mice with and without APA. The liver and spleen permeability to PL, controlled by frli, frs, restrict the amount of drug accessible to the liver and spleen; these properties are not greatly affected by APA status, indicating that, mechanistically, the difference in clearance driven by APA is attributed to APA binding to PL and retention of the resulting APA/PL complexes in the liver via uptake by cells such as LSECs or Kupffer cells. However, the APA need not speed the process of exposing different cells in the liver to PL, which would be a process limited by delivery via blood flow or capillary permeability and reflected in greater sensitivity to drug permeability. This was not the case reflected in the model or the data. Increased total drug uptake in the liver and spleen is further evidenced in comparing the time series for PET signal and AUC values between the cohorts. We have identified that the retention process in the liver, attributed in the model to Kpli and caused physiologically by antibodies cross-linking PL in the liver, is primarily responsible for the increased liver uptake. This is the greatest difference between the mice in the APA+ versus naïve cohorts.
PBPK models have a large number of parameters, and there is always a possibility of multiple optimal parameter ranges consistent with the experimental data. However, we were encouraged by the consistent clustering between mice and the ability of LHS to distinguish naïve and APA+ mice by their non-overlapping ranges for liver and spleen permeation and retention properties, while also revealing greater variability in APA+ mice. Other approaches to parameter estimation, such as Markov Chain Monte Carlo, have been demonstrated for other dynamical systems (Li et al. 2018; Talawar and Aundhakar 2016). Bayesian methods combined with MCMC have been demonstrated for parameter identification in PBPK systems (Krauss et al. 2013), and LHS has been previously employed for quantifying uncertainty in PBPK (Fàbrega et al. 2016). In this study, LHS produced optimized parameter ranges in our PBPK model at a predictable and acceptable computational cost. Furthermore, the non-convex and highly sensitive nature of this optimization problem lends itself to a structured stochastic global algorithm like LHS as opposed to alternative methods. To confirm this, we wrote a gradient descent algorithm and searched for a minimum. We found that gradient descent, initialized in the neighborhood of LHS-determined optimal ranges, indeed converges to optimum parameter values reasonably near the initialization point. However, we also found that using gradient descent with initial values within the naïve ranges used in the initial LHS run may converge to some local minima far above other local minima (Fig. S.5). This demonstrates that when there is uncertainty in the convexity of the model landscape, or if one identifies that the model possesses multiple local minima, LHS can be used as a computationally efficient, semi-random sampling method to identify the global minimum, i.e., the optimal parameter set. We conclude that it is advisable to begin with a comprehensive stochastic search algorithm, such as LHS, and then perhaps subsequently refine within a smaller window using other MCMC, Bayesian, or deterministic methods as presented by An and Choi (2013). Alternatively, one could continue using LHS, or once refined enough, switch to a gradient descent algorithm for subsequent iterations. However, due to inherent variability in our biological system of interest, the potential for non-convexity in the landscape, and noise in the collected data, we assert that LHS is both computationally efficient and most informative of the optimal ranges and values consistent with the experimental data.
Overall, these data-based analyses provide a means to further our understanding of the driving physiological processes behind accelerated drug clearance and to begin to consider strategies for overcoming APA-mediated accelerated clearance.
Availability of Data and Material
All materials are available upon request.
Code Availability
References
Abu Lila AS, Kiwada H, Ishida T (2013) The accelerated blood clearance (ABC) phenomenon: clinical challenge and approaches to manage. J Control Release 172:38–47. https://doi.org/10.1016/j.jconrel.2013.07.026
An D, Choi J-H (2013) Improved MCMC method for parameter estimation based on marginal probability density function. J Mech Sci Technol 27:1771–1779. https://doi.org/10.1007/s12206-013-0428-9
Armstrong JK, Hempel G, Koling S, Chan LS, Fisher T, Meiselman HJ, Garratty G (2007) Antibody against poly(ethylene glycol) adversely affects PEG-asparaginase therapy in acute lymphoblastic leukemia patients. Cancer 110:103–111. https://doi.org/10.1002/cncr.22739
Brown RP, Delp MD, Lindstedt SL, Rhomberg LR, Beliles RP (1997) Physiological parameter values for physiologically based pharmacokinetic models. Toxicol Ind Health 13:407–484. https://doi.org/10.1177/074823379701300401
Cao Y, Jusko WJ (2012) Applications of minimal physiologically-based pharmacokinetic models. J Pharmacokinet Pharmacodyn 39:711–723. https://doi.org/10.1007/s10928-012-9280-2
Davies B, Morris T (1993) Physiological parameters in laboratory animals and humans. Pharm Res 10:1093–1095. https://doi.org/10.1023/A:1018943613122
Edginton AN, Zimmerman EI, Vasilyeva A, Baker SD, Panetta JC (2016) Sorafenib metabolism, transport, and enterohepatic recycling: physiologically based modeling and simulation in mice. Cancer Chemother Pharmacol 77:1039–1052. https://doi.org/10.1007/s00280-016-3018-6
Fàbrega F, Nadal M, Schuhmacher M, Domingo JL, Kumar V (2016) Influence of the uncertainty in the validation of PBPK models: a case-study for PFOS and PFOA. Regul Toxicol Pharmacol 77:230–239. https://doi.org/10.1016/j.yrtph.2016.03.009
Fan J, Zhang X, Zhao L (2017) Utility of physiologically based pharmacokinetic absorption modeling to predict the impact of salt-to-base conversion on prasugrel HCl product bioequivalence in the presence of proton pump inhibitors. AAPS J 19:1479–1486. https://doi.org/10.1208/s12248-017-0116-2
Ganesan LP et al (2012) FcγRIIb on liver sinusoidal endothelium clears small immune complexes. J Immunol 189:4981–4988. https://doi.org/10.4049/jimmunol.1202017
Garg A, Balthasar JP (2007) Physiologically-based pharmacokinetic (PBPK) model to predict IgG tissue kinetics in wild-type and FcRn-knockout mice. J Pharmacokinet Pharmacodyn 34:687–709. https://doi.org/10.1007/s10928-007-9065-1
He H, Liu C, Wu Y, Zhang X, Fan J, Cao Y (2018) A multiscale physiologically-based pharmacokinetic model for doxorubicin to explore its mechanisms of cytotoxicity and cardiotoxicity in human physiological contexts. Pharm Res 35:174. https://doi.org/10.1007/s11095-018-2456-8
He H, Yuan D, Wu Y, Cao Y (2019) Pharmacokinetics and pharmacodynamics modeling and simulation systems to support the development and regulation of liposomal drugs. Pharmaceutics. https://doi.org/10.3390/pharmaceutics11030110
Helton JC, Davis FJ (2003) Latin hypercube sampling and the propagation of uncertainty in analyses of complex systems. Reliab Eng Syst Saf 81:23–69. https://doi.org/10.1016/S0951-8320(03)00058-9
Hershfield MS, Ganson NJ, Kelly SJ, Scarlett EL, Jaggers DA, Sundy JS (2014) Induced and pre-existing anti-polyethylene glycol antibody in a trial of every 3-week dosing of pegloticase for refractory gout, including in organ transplant recipients. Arthritis Res Ther 16:R63. https://doi.org/10.1186/ar4500
Hora SC, Iman RL (1989) Expert opinion in risk analysis: the NUREG-1150 methodology. Nucl Sci Eng 102:323–331. https://doi.org/10.13182/NSE89-A23645
Hsieh NH, Reisfeld B, Bois FY, Chiu WA (2018) Applying a global sensitivity analysis workflow to improve the computational efficiencies in physiologically-based pharmacokinetic modeling. Front Pharmacol 9:588. https://doi.org/10.3389/fphar.2018.00588
Huh Y, Smith DE, Rose Feng M (2011) Interspecies scaling and prediction of human clearance: comparison of small- and macro-molecule drugs. Xenobiotica 41:972–987. https://doi.org/10.3109/00498254.2011.598582
Iman RL, Helton JC, Campbell JE (1981) An approach to sensitivity analysis of computer models: part I—introduction, input variable selection and preliminary variable assessment. J Qual Technol 13:174–183. https://doi.org/10.1080/00224065.1981.11978748
Ishida T, Kiwada H (2008) Accelerated blood clearance (ABC) phenomenon induced by administration of PEGylated liposome. Yakugaku zasshi J Pharm Soc Jpn 128:233–243
Ishida T, Harada M, Wang XY, Ichihara M, Irimura K, Kiwada H (2005) Accelerated blood clearance of PEGylated liposomes following preceding liposome injection: effects of lipid dose and PEG surface-density and chain length of the first-dose liposomes. J Control Release 105:305–317. https://doi.org/10.1016/j.jconrel.2005.04.003
Jones HM, Rowland-Yeo K (2013) Basic concepts in physiologically based pharmacokinetic modeling in drug discovery and development CPT: pharmacometrics & systems. Pharmacology 2:63. https://doi.org/10.1038/psp.2013.41
Kaliss N, Pressman D (1950) Plasma and blood volumes of mouse organs, as determined with radioactive iodoproteins. Proc Soc Exp Biol Med Soc Exp Biol Med 75:16–20. https://doi.org/10.3181/00379727-75-18083
Kleine-Tebbe J et al (2021) Severe allergic reactions to the COVID-19 vaccine: statement and practical consequences. Allergol Sel 5:26–28. https://doi.org/10.5414/ALX02215E
Kozma GT, Shimizu T, Ishida T, Szebeni J (2020) Anti-PEG antibodies: properties, formation, testing and role in adverse immune reactions to PEGylated nano-biopharmaceuticals. Adv Drug Deliv Rev. https://doi.org/10.1016/j.addr.2020.07.024
Krauss M et al (2013) Using Bayesian-PBPK modeling for assessment of inter-individual variability and subgroup stratification. In Silico Pharmacol 1:6–6. https://doi.org/10.1186/2193-9616-1-6
Lang J, Vincent L, Chenel M, Ogungbenro K, Galetin A (2020) Impact of hepatic CYP3A4 ontogeny functions on drug-drug interaction risk in pediatric physiologically-based pharmacokinetic/pharmacodynamic modeling: critical literature review and ivabradine case study. Clin Pharmacol Ther. https://doi.org/10.1002/cpt.2134
Le Merdy M et al (2020) Ocular physiologically based pharmacokinetic modeling for ointment formulations. Pharm Res 37:245. https://doi.org/10.1007/s11095-020-02965-y
Li C, Pei Y, Zhu M, Deng Y (2018) Parameter estimation on a stochastic SIR model with media coverage. Discrete Dyn Nat Soc 2018:3187807. https://doi.org/10.1155/2018/3187807
Lipsky PE, Calabrese LH, Kavanaugh A, Sundy JS, Wright D, Wolfson M, Becker MA (2014) Pegloticase immunogenicity: the relationship between efficacy and antibody development in patients treated for refractory chronic gout. Arthritis Res Ther 16:R60. https://doi.org/10.1186/ar4497
Lutz JD, Mathias A, German P, Pikora C, Reddy S, Kirby BJ (2021) Physiologically-based pharmacokinetic modeling of remdesivir and its metabolites to support dose selection for the treatment of pediatric patients with COVID-19. Clin Pharmacol Ther. https://doi.org/10.1002/cpt.2176
Marino S, Hogue IB, Ray CJ, Kirschner DE (2008) A methodology for performing global uncertainty and sensitivity analysis in systems biology. J Theor Biol 254:178–196. https://doi.org/10.1016/j.jtbi.2008.04.011
McKay MD, Beckman RJ, Conover WJ (1979) A comparison of three methods for selecting values of input variables in the analysis of output from a computer code. Technometrics 21:239–245. https://doi.org/10.2307/1268522
McSweeney MD et al (2018) A minimal physiologically based pharmacokinetic model that predicts anti-PEG IgG-mediated clearance of PEGylated drugs in human and mouse. J Control Release 284:171–178. https://doi.org/10.1016/j.jconrel.2018.06.002
Mohamed M et al (2019) PEGylated liposomes: immunological responses. Sci Technol Adv Mater 20:710–724. https://doi.org/10.1080/14686996.2019.1627174
Mordenti J, Chen SA, Moore JA, Ferraiolo BL, Green JD (1991) Interspecies scaling of clearance and volume of distribution data for five therapeutic proteins. Pharm Res 8:1351–1359. https://doi.org/10.1023/A:1015836720294
Moses (2021). spider_plot (https://github.com/NewGuy012/spider_plot), GitHub. Accessed 4 Feb 2021
Nasu R, Kumagai Y, Kogetsu H, Tsujimoto M, Ohtani H, Sawada Y (2005) Physiologically based pharmacokinetic model for pralmorelin hydrochloride in rats. Drug Metab Dispos Biol Fate Chem 33:1488–1494. https://doi.org/10.1124/dmd.104.001040
Povsic TJ et al (2016) Pre-existing anti-PEG antibodies are associated with severe immediate allergic reactions to pegnivacogin, a PEGylated aptamer. J Allergy Clin Immunol 138:1712–1715. https://doi.org/10.1016/j.jaci.2016.04.058
Pyzik M, Sand KMK, Hubbard JJ, Andersen JT, Sandlie I, Blumberg RS (2019) The Neonatal Fc Receptor (FcRn): a misnomer? Front Immunol. https://doi.org/10.3389/fimmu.2019.01540
Ritschel WA, Vachharajani NN, Johnson RD, Hussain AS (1992) The allometric approach for interspecies scaling of pharmacokinetic parameters. Comp Biochem Physiol Part C Comp Pharmacol 103:249–253. https://doi.org/10.1016/0742-8413(92)90003-P
Stader F, Battegay M, Marzolini C (2021) Physiologically based pharmacokinetic modelling to support the clinical management of drug-drug interactions with bictegravir. Clin Pharmacol Ther. https://doi.org/10.1002/cpt.2221
Talawar AS, Aundhakar UR (2016) Parameter estimation of SIR epidemic model using MCMC methods. Glob J Pure Appl Math 12:1299–1306
Tsamandouras N, Rostami-Hodjegan A, Aarons L (2015) Combining the ‘bottom up’ and ‘top down’ approaches in pharmacokinetic modelling: fitting PBPK models to observed clinical data. Br J Clin Pharmacol 79:48–55. https://doi.org/10.1111/bcp.12234
Ueno T, Miyajima Y, Landry I, Lalovic B, Schuck E (2021) Physiologically based pharmacokinetic modeling to predict drug interactions of lemborexant with CYP3A inhibitors. CPT Pharmacomet Syst Pharmacol. https://doi.org/10.1002/psp4.12606
Walsh C et al (2016) Development of a physiologically based pharmacokinetic model of actinomycin D in children with cancer. Br J Clin Pharmacol 81:989–998. https://doi.org/10.1111/bcp.12878
Worm M et al (2021) Practical recommendations for the allergological risk assessment of the COVID-19 vaccination: a harmonized statement of allergy centers in Germany. Allergol Sel 5:72–76. https://doi.org/10.5414/ALX02225E
Yang Q, Lai SK (2015) Anti-PEG immunity: emergence, characteristics, and unaddressed questions. Wiley Interdiscip Rev Nanomed Nanobiotechnol 7:655–677. https://doi.org/10.1002/wnan.1339
Yuan D, He H, Wu Y, Fan J, Cao Y (2019) Physiologically based pharmacokinetic modeling of nanoparticles. J Pharm Sci 108:58–72. https://doi.org/10.1016/j.xphs.2018.10.037
Acknowledgements
The authors would like to thank Morgan McSweeney for meaningful discussions contributing to this work. We would also like to thank the staff at the Biomedical Research Imaging Center and Animal Studies Core at the University of North Carolina at Chapel Hill.
Funding
This work was supported by The David and Lucile Packard Foundation (2013-39274, SKL), National Institutes of Health (T32-HL069768, AMT; HL141934, SKL; R35GM119661, YC), P.E.O. International (AMT), an Eshelman Institute of Innovation award (SKL), and the National Science Foundation (DMS-1664645, DMS-1816630, CISE-1931516, MGF).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection, and analysis were performed by Anne M. Talkington and Timothy Wessler. The first draft of the manuscript was written by Anne M. Talkington and Timothy Wessler, and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors have no conflict of interest to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Talkington, A.M., Wessler, T., Lai, S.K. et al. Experimental Data and PBPK Modeling Quantify Antibody Interference in PEGylated Drug Carrier Delivery. Bull Math Biol 83, 123 (2021). https://doi.org/10.1007/s11538-021-00950-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11538-021-00950-z