
Abstract
Inhibitors of Bruton’s tyrosine kinase (BTK), a major kinase in the B-cell receptor (BCR) signaling pathway, mediating B-cell proliferation and apoptosis, have substantially altered the management, clinical course, and outcome of patients with B-cell malignancies. This is especially true for patients with previously limited treatment options due to disease characteristics or coexisting diseases. Ibrutinib was the first orally available, nonselective and irreversible inhibitor of BTK approved for the treatment of patients with various B-cell malignancies. Newer and more selective BTK inhibitors are currently in clinical development, including acalabrutinib, which is currently US FDA approved for previously treated mantle cell lymphoma. Significant efforts are underway to investigate the optimal combinations, timing, and sequencing of BTK inhibitors with other regimens and targeted agents, and to capitalize on the immunomodulatory modes of action of BTK inhibitors to correct tumor-induced immune defects and to achieve long-lasting tumor control. This review describes the major milestones in the clinical development of BTK inhibitors in chronic lymphocytic leukemia and other B-cell malignancies, highlights the most recent long-term follow-up results, and evaluates the role of BTK inhibitors and their combination with other agents in B-cell malignancies and other indications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Bruton’s tyrosine kinase (BTK) inhibitors are an effective treatment strategy and have been paradigm shifting for a number of B-cell malignancies. |
Specific side effects with ibrutinib, including arrhythmias and bleeding, have been challenging in clinical practice and occasionally require discontinuation of the drug or addition of other supportive care. |
Second-generation irreversible BTK inhibitors are currently in development and may offer a safer alternative for some patients. |
1 Introduction
In the last 2 decades, the hematology community has witnessed several paradigm shifts in the treatment of patients with B-cell malignancies and especially chronic lymphocytic leukemia (CLL). First, the addition of the cluster of differentiation (CD)-20-targeting monoclonal antibody rituximab to chemotherapy [fludarabine and cyclophosphamide (FCR)] improved progression-free survival (PFS) and overall survival (OS) in previously untreated CLL patients. Later, this regimen was found to lead to extremely durable remissions in a subset of patients, especially those without TP53 abnormalities and with mutated immunoglobulin heavy chain variable (IgHV) gene status [1,2,3]. A direct comparison between frontline FCR and a combination of bendamustine with rituximab (BR) demonstrated longer median PFS after FCR treatment, especially in IgHV-unmutated patients [4]. However, FCR also led to higher rates of adverse events, such as cytopenias, infection and potentially secondary malignancies, especially in older patients. Based on these findings, FCR remained the front-line standard of care for younger and fit CLL patients with favorable prognostics. For elderly patients and those with co-existing morbidities, BR and the combination of chlorambucil with obinutuzumab, a type-2 CD20 antibody, were widely considered reasonable options [5]. These approaches were again revolutionized with the advent of novel agents targeting components of the B-cell receptor signaling pathway and inhibitors of the BCL2 apoptotic pathway, especially in patients who were too frail for conventional chemoimmunotherapy or those with poor prognostic and high-risk features of disease. Risk categories in CLL are generally defined by genetic criteria, response, and duration of response (DOR) to previous treatment [6]. Patients with TP53 abnormalities, refractoriness to purine analog-based regimens, or very short response and lack of complete remission (CR) after prior chemoimmunotherapy are known to have the highest risk of adverse outcome with conventional chemoimmunotherapy and require alternative treatment options. While allogeneic transplantation was commonly considered the only available potentially curative treatment option for such high-risk CLL patients, these patients are now generally treated with the oral Bruton’s tyrosine kinase (BTK) inhibitor ibrutinib [7, 8]. In addition to CLL, ibrutinib has added significant options to the treatment of patients with previously treated mantle cell lymphoma (MCL), Waldenström’s macroglobulinemia (WM), and marginal zone lymphoma (MZL). To capitalize on the efficacy of ibrutinib, several second-generation BTK inhibitors have been developed. Among these, the highly selective BTK inhibitor acalabrutinib has demonstrated significant activity in patients with CLL and MCL. This review briefly summarizes the biological function of BTK in B cells and other immune cells, describes the major milestones in the clinical development of BTK inhibitors in CLL and other B-cell malignancies, highlights the most recent long-term follow-up results and critically evaluates the role of BTK inhibitors and their combination with other agents in B-cell malignancies and other indications.
2 Bruton’s Tyrosine Kinase (BTK) is an Essential Component of B-Cell Receptor Signaling and Mediates Effector Functions in Non-Malignant Immune and Stromal Cells
The physiological role of BTK in mediating B-cell receptor (BCR) signaling and how this is altered in B-cell malignancies has been extensively described in other reviews [9, 10]. Briefly, engagement of the BCR by antigen induces the activation of a network of kinases and phosphatases that tune and amplify the incoming signal. Tyrosine kinases such as LYN, SYK and BTK and various adapter molecules mediate the activation of downstream signaling pathways that are essential for B-cell proliferation and apoptosis, such as phosphoinositide-3-kinase (PI3K)/protein kinase B (AKT), nuclear factor (NF)-ĸB and extracellular-signal-regulated kinase/mitogen-activated protein kinase [extracellular signal–regulated kinases (ERK)/MAPK]. CLL cells have been shown to exhibit heterogeneous but constitutively active BCR signaling, as a result of both a biased BCR repertoire and chronic antigenic stimulation, resulting in the over-activation of pro-proliferative and anti-apoptotic pathways. It is well-established that CLL pathogenesis is also determined by various and complex interactions with non-malignant immune cells such as T, myeloid and NK cells and stromal components [11]. Importantly, BTK and other Tec kinase family members such as Tec, ITK, RLK and BMX are essential regulators of key immune functions in both innate and adaptive immune cells and mediate signaling via G-protein-coupled, integrin, cytokine/chemokine, Toll-like, T-cell and Fcγ receptors [12]. As a result, BTK inhibition in CLL has been described by various studies to alter cellular migration and homing abilities, T- and myeloid-cell differentiation and activation, and antimicrobial and other innate immunity functions such as phagocytosis and pro-inflammatory cytokine production [13,14,15,16]. These findings emphasize that pharmacologic inhibition of BTK holds the potential to not only control aberrant BCR signaling in malignant cells but also correct CLL-induced immune dysfunction. This is of utmost clinical relevance, as global immune dysfunction leading to increased rates of infections is the major cause of increased morbidity and mortality especially among patients with CLL [17].
3 Landmark Trials in the Clinical Development of Single-Agent BTK Inhibitors for the Treatment of B-Cell Malignancies
3.1 Currently Approved Indications of Ibrutinib
The first-in class oral BTK inhibitor ibrutinib (trade name IMBRUVICA®) is currently approved for use as a single agent for patients with CLL/small lymphocytic leukemia (SLL), MCL after at least one prior therapy, WM, MZL requiring systemic therapy and after previous treatment with at least one anti-CD20-based regimen, and chronic graft versus host disease (cGVHD) after failure of one or more lines of systemic therapy [18].
3.2 Ibrutinib in Relapsed/Refractory Hematological Malignancies
During the first phase I trial enrolling patients with relapsed/refractory (R/R) non-Hodgkin lymphomas (NHL), CLL or WM for whom at least one previous therapy had failed, ibrutinib was administered orally once daily at 1.25, 2.5, 5, 8.3, or 12.5 mg/kg on a 35-day cycle, with 28 days on and 7 days off treatment, or continuously at 8.3 mg/kg or 560 mg per day until disease progression, unacceptable toxicity or the decision to end therapy [19]. Treatment was generally well-tolerated, even with prolonged dosing, and objective response rates were observed in 60% of patients, encouraging further clinical development.
3.3 Ibrutinib in Marginal Zone Lymphoma
In patients with previously treated MZL, the efficacy and safety of single-agent ibrutinib 560 mg daily was examined in a multicenter, open-label, phase II study [20]. The overall response rate (ORR) was 48% and median PFS was 14.2 months, with a favorable benefit-risk profile, supporting the use of ibrutinib in a patient cohort where no gold-standard treatment exists.
3.4 Ibrutinib in Waldenström’s Macroglobulinemia
In 63 patients with WM who had received at least one previous line of treatment, ibrutinib 420 mg daily was highly active and associated with an ORR of 91%, especially in patients with MYD88 and CXCR4 mutations, which are recognized drivers of malignant cell growth via increased BTK signaling [21]. In a population of 31 patients with WM with rituximab-refractory disease, the ORR was 90% and the estimated 18-month OS rate was 97%, highlighting single-agent ibrutinib as a new treatment approach in this disease [22]. A recently published study prospectively evaluated single-agent ibrutinib in symptomatic, treatment-naïve patients with WM and mutations in MYD88 and/or CXCR4 [23]. The ORR was 100% and 18-month PFS was 92%, demonstrating that front-line ibrutinib might be an effective treatment option in patients with symptomatic WM and MYD88 and CXCR4 mutations.
3.5 Ibrutinib in Mantle Cell Lymphoma (MCL)
The international, multicenter, open-label, phase II trial that led to the accelerated approval of ibrutinib in patients with R/R MCL demonstrated a high ORR of 68%, with 21% complete response rates and a median DOR of 17.5 months [24]. A recent analysis pooled the findings from several available trials of patients with R/R MCL after 3.5 years of follow-up and reported an ORR of 69.7% [25]. Of note, 26.5% of patients achieved CR, especially if just one prior line of treatment had been administered.
3.6 Ibrutinib in Chronic Lymphocytic Leukemia (CLL)
Results from the first phase Ib/II study of ibrutinib in both treatment-naïve (TN) elderly (aged ≥ 65 years) and R/R CLL patients yielded acceptable toxicities and similar ORRs in patients receiving 420 mg and 840 mg [26]. This was particularly remarkable as elderly patients with CLL represent a unique cohort with an unmet clinical need, as conventional treatment approaches are often too toxic [27]. In the cohort of 31 previously untreated CLL/SLL patients aged ≥ 65 years (median 71), ibrutinib produced only moderate toxicity and an objective response in 89% [28]. Importantly, CR rates increased over time to 29% in TN patients and to 10% in R/R patients. In TN patients, median PFS was not reached, and the 5-year PFS was 92%. In R/R patients, PFS was 51 months, and the 5-year PFS was 44%. Of note, a shorter median PFS of 26 months was observed for patients with del(17p) and in those with unmutated IgHV (43 months), indicating that ibrutinib might not be able to overcome the adverse prognostic role of known high-risk cytogenetic or biological factors. However, subsequently published clinical trials focusing on specific outcomes of single-agent ibrutinib treatment in patients with TP53 abnormalities supported the use of single-agent ibrutinib in patients with CLL with high-risk cytogenetic characteristics in a front-line and relapse treatment setting. A phase II single-arm study evaluated the safety and activity of ibrutinib in previously untreated and R/R CLL patients with TP53 aberrations treated at the National Institutes of Health (NIH) Clinical Center [29]. Patients received ibrutinib 420 mg once daily until disease progression or the occurrence of intolerable toxicities. After a median follow-up of 24 months, the ORR was 97% in TN and 80% in R/R patients. With extended 5-year follow-up, depth of response improved over time, and 5-year PFS was 74% in TN and 19.4% in R/R CLL patients, with an OS rate of 85% and 54%, respectively [30]. Another multicenter, international, open-label, single-arm study confirmed high ORR and a favorable toxicity profile in 145 previously treated patients with CLL/SLL with del(17p) receiving ibrutinib 420 mg once daily [31]. After a median follow-up of 27.6 months, the investigator-assessed ORR was 83%, and 24-month PFS and OS were 63% and 75%, respectively. Together, the response and PFS rates in these trials were substantially higher than those reported after treatment with FCR [1], leading to the integration of BTK inhibitors into treatment recommendations for patients with TP53 abnormalities. An integrated analysis of 230 patients with R/R with TP53 abnormalities treated within the several trials of ibrutinib single agents described [26, 31,32,33] reported an ORR of 85% and estimated 30-month PFS and OS rates of 57% and 69%, respectively, confirming that ibrutinib surpasses other currently available treatment regimens in this high-risk patient population [34].
3.7 Phase III Clinical Trials of Single-Agent Ibrutinib in MCL and CLL
The efficacy of ibrutinib as single-agent treatment has also been assessed in several phase III clinical trials. Patients with R/R MCL were randomized to receive either oral ibrutinib 560 mg or intravenous temsirolimus [35]. Patients with MCL treated with ibrutinib had significantly improved PFS, lower toxicity and fewer discontinuations of study medication due to adverse events than those treated with temsirolimus. Several major phase III trials of single-agent ibrutinib in CLL have been reported. The RESONATE trial randomized patients with R/R CLL/SLL to receive daily ibrutinib or the anti-CD20 antibody ofatumumab [32]. After a median follow-up of 9.4 months, ibrutinib significantly improved ORR, PFS and OS, even in patients with del(17p) and those with resistance to purine analogues. The RESONATE-2 trial randomized 269 previously untreated CLL/SLL patients aged ≥ 65 years without del(17p) to treatment with single-agent ibrutinib or chlorambucil [36]. After a median follow-up of 18.4 months, ibrutinib significantly improved ORR, PFS and OS versus chlorambucil. Recently published extended follow-up data demonstrated sustained responses and PFS benefits in the ibrutinib treatment arm as well as a substantial increase in CR rates with longer duration of treatment [37]. The most recently published phase III ALLIANCE trial compared the efficacy of ibrutinib versus BR versus ibrutinib combined with rituximab in treatment-naïve CLL patients ≥ 65 years, including those with high-risk features [38]. Similar to previous studies, the addition of ibrutinib significantly improved PFS and ORR compared with chemoimmunotherapy with BR alone, whereas there were no significant differences in PFS between the two ibrutinib-containing treatment arms. A summary of phase III clinical trials in CLL is provided in Table 1.
4 Finding the Most Suitable Partner: Landmark Clinical Trials of Ibrutinib Combinations
4.1 Ibrutinib Plus Anti-CD20 Antibodies
Several attempts have been made to improve the efficacy of ibrutinib single-agent treatment by adding another agent for synergistic or complementary action. The first report of ibrutinib combination therapy was published in 2014 [39]. This phase II trial evaluated the combination of ibrutinib with rituximab in 40 patients with high-risk CLL, demonstrating that the addition of rituximab led to 18-month PFS rates of 78% in all patients and 73% in those with TP53 abnormalities. After extended follow-up (median 47 months), median PFS was 45 months and median OS was not reached [40]. The ORR was 95%, with 23% of patients attaining CR. Of note, PFS was shorter in patients with del(17p). However, data from the recently published ALLIANCE phase III clinical trial demonstrated that the addition of rituximab to ibrutinib did not improve PFS or ORR over single-agent ibrutinib in TN older CLL patients (2-year PFS ibrutinib 87% vs. ibrutinib/rituximab 88%; ORR ibrutinib 93%; 95% confidence interval [CI] 88–96; ibrutinib/rituximab 94%; 95% CI 89–97) [38]. Of note, the percentage of patients with undetectable minimal residual disease (MRD) was significantly higher with chemoimmunotherapy than with any of the ibrutinib-containing regimens. Moreover, BR chemoimmunotherapy was associated with higher rates of grade 3 or higher hematologic adverse events but lower rates of grade 3 or higher non-hematologic adverse events such as atrial fibrillation (AF) and hypertension than were ibrutinib-containing regimens. In younger TN patients with CLL aged < 70 years and excluding those with del(17p), ibrutinib and rituximab were recently described as resulting in superior PFS (hazard ratio (HR) 0.352; 95% CI 0.223–0.558; p < 0.0001) and OS (HR 0.168; 95% CI 0.053–0.538; p = 0.0003) rates compared with FCR chemoimmunotherapy, challenging the current gold-standard treatment for younger fit patients with CLL [41]. Moreover, grade 3 and 4 treatment-related adverse events such as neutropenia and infectious complications occurred more frequently in FCR-treated patients (p = 0.0042).
The combination of ibrutinib with rituximab was also assessed in patients with WM and MCL. In 150 symptomatic patients with both TN and pre-treated WM, ibrutinib + rituximab yielded a significant PFS benefit over placebo + rituximab (30-month PFS 82 vs. 28%), independent of MYD88 or CXCR4 genotype [42]. While ibrutinib + rituximab produced superior responses, it was also associated with a higher rate of AF and hypertension adverse events. Another trial treated 50 patients with R/R MCL with continuous ibrutinib 560 mg/day and rituximab once weekly for the first cycle, then once per month until cycle 8, and then once every other cycle for up to 2 years [43]. At a median follow-up of 16.5 months, 88% of patients achieved an objective response, and 44% a CR. Adverse events such as grade 3 AF and grade 4 diarrhea and neutropenia led to discontinuation of therapy in only a small number of patients, supporting further investigation of this combination in a phase III trial.
Ibrutinib was also investigated in combination with the anti-CD20 antibody ofatumumab in 71 R/R patients with CLL/SLL, prolymphocytic leukemia, or Richter’s transformation [44]. This trial used three different administration sequences to evaluate the effect of ibrutinib lead-in versus concurrent start versus ofatumumab lead-in. Interestingly, ORR and 12-month PFS were highest among those with ibrutinib lead-in, with generally good tolerability and clinical activity in all administration schedules. The efficacy of a combination of ibrutinib with the anti-CD20 antibody obinutuzumab (I-G) versus chlorambucil + obinutuzumab (clb-G) for TN patients with CLL/SLL was recently examined in the phase III iLLUMINATE trial [45]. I-G significantly prolonged PFS compared with clb-G regardless of high-risk genomic features, led to an ORR of around 90% and produced MRD-negativity in 35% of patients (vs. 25% clb-G-treated patients). A summary of phase III clinical trials in CLL is provided in Table 1.
4.2 Ibrutinib Plus Chemoimmunotherapy
The international, double-blind phase III HELIOS trial compared ibrutinib combined with bendamustine and rituximab (I-BR) versus placebo and BR in 578 previously treated patients with CLL/SLL [46]. Of note, patients with del(17p) were excluded because of their known intrinsic resistance to BR therapy. At a median follow-up of 17 months, PFS was significantly improved in the ibrutinib group compared with the placebo group, and 18-month PFS was 79% in the ibrutinib and 24% in the placebo group, with no excess toxicity, suggesting that the addition of ibrutinib to BR resulted in significant improvements in outcome over chemoimmunotherapy alone. Recently published updated results (median follow-up 34.8 months) confirmed improved survival outcomes and deepening of responses with I-BR compared with BR alone, with 36-month PFS rates of 68 versus 14% [47]. As described, ibrutinib alone and ibrutinib + rituximab were compared with BR only in the phase III ALLIANCE trial, and showed significant improvements in PFS and ORR in ibrutinib-containing regimens [38]. While MRD-negative responses were significantly less frequent in ibrutinib treatment arms in the ALLIANCE trial, MRD-negative response rates were 26% for I-BR versus 6% for BR in the HELIOS trial, suggesting that synergistic action between BR-chemoimmunotherapy and ibrutinib is needed to improve molecular remissions. A summary of phase III clinical trials in CLL is provided in Table 1.
4.3 Ibrutinib Plus Other Targeted Agents
Other regimens that have already been reported in the literature include ibrutinib plus rituximab plus the immunomodulatory agent lenalidomide in patients with R/R CLL [48], which did not appear superior to prior reports of the rituximab + lenalidomide combination or single-agent ibrutinib. In 50 patients with R/R MCL, ibrutinib was administered with rituximab and lenalidomide during an induction phase of 12 cycles of 28 days, followed by maintenance with ibrutinib and rituximab only (PHILEMON trial) [49]. At a median follow-up of 17.8 months, the ORR was 76%, including 56% of patients with CR. Toxicities included grade 3–4 adverse neutropenia, infections and cutaneous toxicity, and three treatment-related deaths were reported.
Findings of a completely chemotherapy-free regimen consisting of ibrutinib monotherapy for three cycles followed by the addition of venetoclax, a selective, orally bioavailable inhibitor of BCL2, were recently reported for 80 patients with treatment-naïve CLL [50]. After 12 months of combination treatment, CR rates were > 90%, even in older patients and those with high-risk features, rates of molecular response were also high. A similar approach was used in the CLARITY trial, where venetoclax was added after 8 weeks of ibrutinib monotherapy for the treatment of patients with R/R CLL and produced a high rate of MRD eradication, especially with a longer duration of combination treatment [51]. A recently published study reported the combination of ibrutinib and venetoclax in 24 patients with MCL with high-risk features [52]. Venetoclax was added in weekly increasing doses to 400 mg per day after 4 weeks of ibrutinib monotherapy. This yielded an overall CR rate of 71% and MRD clearance by flow cytometry in 67% of patients. Common side effects were generally limited, suggesting that this combination leads to improved outcomes over conventional or single-agent ibrutinib treatment strategies. Another BCL2/BTK inhibition combination currently in clinical development for patients with TN and R/R CLL is obinutuzumab, ibrutinib and venetoclax, which led to a high ORR of 92% and achieved CR in almost half of all treated patients [53, 54].
Dual B-cell receptor pathway blockade was recently reported in an ongoing phase I study of ibrutinib with umbralisib, a PI3K-δ inhibitor (PI3K-δi), in 44 patients with R/R CLL and MCL [55]. While no dose-limiting toxicities were observed, additional follow-up will be needed to determine the clinical efficacy and durability of responses achieved with this regimen. The combination of umbralisib and ibrutinib with ublituximab, a third-generation anti-CD20 monoclonal antibody with enhanced antibody-dependent cytotoxicity, resulted in an ORR of 84% in 44 patients with R/R B-cell malignancies, supporting further investigation of this chemotherapy-free approach [56]. Combinations of ibrutinib with anti-CD19 chimeric antigen-receptor T (CAR-T) cells have also been investigated in patients with CLL [57], leading to improved responses and enhanced engraftment, probably as a result of correction of disease-induced T-cell defects.
5 Commonly Reported Side Effects and Toxicities of Ibrutinib Treatment
5.1 Early Transient Lymphocytosis
Due to an efflux of cells from lymphoid tissues into blood that is mediated via BTK- and VLA-4 dependent adhesion mechanisms and increased CLL apoptosis, early transient lymphocytosis is observed in nearly all patients treated with ibrutinib [58, 59]. Heavy water-labelling studies in patients undergoing ibrutinib therapy demonstrated that ibrutinib directly reduced the birth rate of malignant clones while increasing their death rates, both in peripheral blood and in tissue compartments [60]. Persistent lymphocytosis during ibrutinib therapy (i.e., lasting > 12 months) was associated with activated downstream mediators of BCR signaling such as ERK, Mitogen-activated protein kinase1/2 and AKT, whereas cells were unresponsive to ex vivo BCR stimulation and carried disease-specific features that were also already present at baseline [61]. Importantly, PFS was not inferior when compared with patients lacking persistent lymphocytosis, suggesting persistence of a quiescent clone rather than evolution and outgrowth of a clone with increased malignant potential.
5.2 Arrhythmias
One of the most commonly observed side effects of ibrutinib therapy are arrhythmias, especially AF, which has been reported in up to 16% of patients. In a pooled analysis of 1505 patients with CLL or MCL enrolled in randomized clinical trials, the incidence of AF was 6.5% at 16.6-month and 10.4% at 36-month follow-up, with an estimated cumulative incidence of 14% [62]. Independent risk factors for AF were ibrutinib treatment, prior history of AF and age ≥ 65 years. Another study of 582 patients with hematologic malignancies treated with ibrutinib reported an estimated cumulative incidence of 10% at 24 months and a median time to onset of AF of 7.6 months [63]. A prior history of AF and a high Framingham Heart Study AF risk score (> 20) were significant risk factors for development of AF. A recently published prospective evaluation of 43 patients with CLL undergoing ibrutinib treatment observed new-onset AF in 16% of patients, which was correlated with the presence of pre-existing cardiac conditions and risk factors and higher left atrial diameter and area [64]. Atrial arrhythmias do not always necessitate drug discontinuation, and many patients can continue to safely receive the drug with rate or rhythm control along with anticoagulation reserved for patients at high risk of stroke. However, as more irreversible BTK inhibitors are approved that have less risk of AF, switching patients with AF to an alternative BTK inhibitor may become the preferred strategy.
More concerning than the atrial arrhythmias, ibrutinib is also associated with a markedly increased risk of ventricular arrhythmia events, even after accounting for baseline cardiovascular disease [65]. For this reason, patients who report palpitations on ibrutinib or have documented ventricular arrhythmias should discontinue ibrutinib in favor of an alternative agent, unless under close supervision by a cardiologist.
5.3 Bleeding-Related Adverse Events
Another commonly occurring adverse event is increased risk of minor hemorrhage, which was reported in about half of all patients with CLL treated in the NIH trial [66] and in ibrutinib-treated patients with MCL [67]. The cumulative incidence of bleeding plateaued after 6 months of treatment, and pre-treatment parameters associated with a significantly increased risk of bleeding-related events included prolonged epinephrine closure time as well as lower levels of von Willebrand factor activity and factor VIII. A meta-analysis of published clinical trials of patients treated with ibrutinib demonstrated an almost threefold higher overall bleeding risk, but a major bleeding risk comparable to that with other treatments [68]. Quantitative assessments of platelet function using tools such as ristocetin-induced platelet aggregation (RIPA) have been proposed as clinical tools to monitor bleeding tendencies in patients receiving BTK-inhibitor treatment [69]. Although this risk of bleeding remains a significant clinical concern, avoidance of warfarin and judicious use of other blood thinners in patients receiving ibrutinib has decreased significant bleeding rates in clinical trials.
5.4 Risk of Infections
Chronic lymphocytic leukemia is generally associated with an increased risk of infections, and several studies indicate that this risk remains high during the first 6 months of ibrutinib treatment [70, 71]. While most opportunistic infections are not seen at high frequency with ibrutinib, invasive aspergillus has occurred more frequently than expected, likely due to the role of BTK in the macrophage response necessary for aspergillus clearance [72, 73]. Early infections were the most common reason of non-relapse treatment discontinuation in a cohort of 232 patients treated at our institution [74]. Of note, patients with an increase in serum IgA of ≥ 50% from baseline to 12 months have been reported to have a significantly lower rate of infections, indicating that long-term ibrutinib treatment fosters recovery of humoral immune function [75]. No specific prophylaxis is currently recommended with ibrutinib, but ongoing studies will continue to assess the risk of specific infections and may lead to guidelines in the future.
6 Factors Determining Response to and Outcome of Ibrutinib Treatment
6.1 Clinical and Disease-Specific Predictors of Response
Maturing follow-up data from several clinical trials suggests that CR rates generally improve with extended duration of treatment. A recently published pooled analysis of two clinical trials analyzed the baseline factors predicting CR in 327 patients with CLL/SLL [76]. Factors that predicted an increased likelihood of CR in a multivariate analysis were lack of previous therapies and absence of bulky disease. Other studies identified beta2-microglobulin normalization within the first 6 months of ibrutinib treatment as a predictor of PFS after adjusting for the effects of pre-treatment beta2-microglobulin levels, R/R disease and presence of del(17p) [77]. It also appears that outcomes are directly affected by ibrutinib dose adherence. Among patients treated in the RESONATE trial, treatment interruptions of at least 8 consecutive days was associated with shorter median PFS, although treatment was continued at the original dose of 420 mg in > 90% of patients [78]. Moreover, several studies found that poor PFS and OS were associated with complex karyotype and near tetraploidy [79, 80].
6.2 Development of Resistance to BTK Inhibition
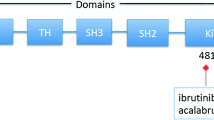
A rare but serious and clinically challenging outcome of ibrutinib treatment is development of resistance to irreversible inhibition that eventually leads to relapse. Massive parallel sequencing at baseline and at time of relapse on samples from six patients with acquired resistance to ibrutinib therapy and from nine patients with CLL with prolonged lymphocytosis identified a cysteine-to-serine mutation in BTK (C481S) at the ibrutinib-binding site in five ibrutinib-resistant patients [81]. In addition, distinct mutations (R665W and L845F) in the immediate downstream effector phospholipase C γ2 (PLCγ2) were identified in two ibrutinib-resistant patients that led to autonomous B-cell-receptor activity bypassing the BTK pathway [74, 81, 82]. Ibrutinib-naïve patients with CLL consistently lack BTK or PLCG2 mutations, and several studies have confirmed that clones with these mutations arise months before relapse, suggesting that monitoring their occurrence and evolution might predict future relapse and guide alternative treatment decisions [83, 84]. Deep sequencing of serial samples of patients with high-risk CLL with complex cytogenetics revealed that clinical resistance might also be driven by non-BTK/PLCG2-mutated subclones that are already present before ibrutinib but are selected and expand during treatment [85]. Serial exome and transcriptome sequencing for a larger cohort of 61 ibrutinib-treated patients with CLL demonstrated clonal shifts in 31% of patients during the first year of therapy, which was associated with adverse outcomes [86]. This study also identified additional previously undescribed mutations in BTK and PLCG2 and alterations such as del(8p), CARD11 G126D mutation and ITPKB somatic substitution in 17 patients at the time of progression. Of note, these mutations have also been described as drivers of genomic evolution in diffuse large BCL (DLBCL) [87, 88]. Resistance mechanisms also occur in ibrutinib-treated patients with WM, which are largely driven by subclonal BTK Cys481 variants and BTK mutations associated with CXCR4 mutations [89]. Interestingly, patients with WM without BTK C481S or CXCR4 mutations exhibited ibrutinib resistance through Bcl2 and AKT upregulation [90]. In MCL, ibrutinib resistance is mainly driven by interactions between malignant cells and their non-malignant environment that promote alternative signaling via PI3K-AKT-mammalian target of rapamycin (mTOR) and integrin-β1 [91]. Together, while BTK and PLCG2 mutations are clearly associated with the development of resistance to ibrutinib in CLL, other mechanisms and pathways also contribute, especially in diseases other than CLL. Further studies are needed to evaluate the causal and temporal relationship of these mechanisms to understand how they can be inhibited, preferably prior to clinical relapse [92].
6.3 Management and Outcome of Patients after Ibrutinib Discontinuation
The management of patients who no longer respond to ibrutinib or for whom this therapy fails is very challenging, especially for those who develop resistance via Richter’s transformation [35, 74, 83, 84, 93]. Richter’s transformation is the transformation of CLL to aggressive lymphoma, and several studies have reported that this generally occurs early during the first 12 months of ibrutinib treatment, affecting around 5% of patients [40, 74, 83, 84]. Current recommendations on treatment algorithms and clinical management are largely focused on identifying suitable patients for allogeneic transplant or treatment with other targeted agents [7, 94, 95]. Several salvage strategies are currently being explored, and it appears that treatment with venetoclax has effective and durable clinical activity and might serve as a bridging strategy for transplant [96, 97]. A recent interim report of a phase II trial of venetoclax in 127 patients with CLL progressing after ibrutinib demonstrated an ORR of 65% [96]: 19% of patients died, with progression being the cause of death in about half of these patients. Durable and deep remissions were also observed in 24 patients with CLL treated with CD19 CAR-T cells [98]. After lymphodepleting chemotherapy, anti-CD19 CAR-T cells were administered at different dose levels. At 4 weeks after CAR-T cell infusion, the ORR was 71%. Remarkably, all patients who responded according to International Workshop on CLL criteria and in whom malignant immunoglobulin H (IgH) clones were eradicated were free of disease after 6.6 months of follow-up. Additional strategies such as CD19/CD3-bispecific antibodies [99] and inhibitors of immune checkpoints [100], to name a few, are currently in preclinical and clinical development, but a comprehensive discussion is outside the scope of this article.
7 Second-Generation Irreversible BTK Inhibitors
Given the irreversible inhibition of non-BTK kinase targets by ibrutinib, several more selective inhibitors of BTK have been developed to increase the safety and efficacy of this treatment. The highly selective BTK inhibitor acalabrutinib (ACP-196) led to a 95% ORR in all 61 included patients with R/R CLL [101]. While it was also highly effective in patients with CLL with del(17p), CRs were limited. In 124 patients with R/R MCL, oral acalabrutinib 100 mg twice daily led to an ORR of 81% and CR in 40% of patients after a median follow-up of 15.2 months with a favorable safety profile [102]. As a result, acalabrutinib (trade name CALQUENE®) has been granted accelerated approval for patients with MCL who have received at least one prior therapy [103]. Several combination strategies are currently being explored in preclinical models, which include combination with other targeted agents such as venetoclax [104] or PI3K-δ inhibition [105]. Another highly selective BTK inhibitor with promising clinical activity is ONO/GS-4059 (tirabrutinib). Phase I data demonstrated very rapid objective responses in 96% of patients in the CLL and in 92% in the MCL group [106]. While much lower responses were seen in non-germinal center DLBCL, severe hematologic toxicity was rare and transient in all 90 enrolled patients. With extended 3-year follow-up, median PFS and OS rates of 38.5 and 44.9 months were reported, and no cases of Richter transformation had occurred in patients with CLL, contrasting the clinical patterns observed with ibrutinib [107]. Results of a phase I trial of BGB-3111 (zanubrutinib) in patients with R/R B-cell malignancies, so far only presented in abstract form, demonstrated an ORR of 90%, and no unanticipated safety concerns were reported. Importantly, this drug is shown to have excellent occupancy of BTK in both the peripheral blood and the lymph nodes [108]. In contrast, CC-292 yielded a much lower ORR of 53% and substantially less durability in patients with CLL/SLL, with the underlying reasons not completely understood [109]. Various clinical trials are underway in CLL and other B-cell malignancies to directly compare different BTK inhibitors.
8 How to Capitalize on the Success of BTK Inhibitors
8.1 In Currently Approved Indications
As discussed, clinical studies have largely been focused on the efficacy and long-term outcomes of single-agent ibrutinib and ibrutinib combination treatments in patient cohorts with distinct risk categories to overcome the lack of durable responses or high toxicities of conventional therapies. Another important question that warrants additional clinical studies is how to sequence BTK inhibitors with other agents. A retrospective analysis of 683 patients with CLL treated with ibrutinib, idelalisib or venetoclax at different institutions reported significantly better PFS in patients treated with ibrutinib as first kinase inhibitor compared with idelalisib, both in a relapsed and TN setting and in patients with del(17p) and complex cytogenetics [97]. Importantly, upon failure of first kinase inhibitor treatment, chemoimmunotherapy was inferior to both alternative kinase inhibitor and venetoclax. Prospective clinical trials testing sequencing strategies to optimize these treatment algorithms are currently being evaluated within the CLL2-BXX phase II trials by the German CLL Study Group (GCLLSG) [110]. These trials are evaluating sequential regimens of bendamustine followed by obinutuzumab and ibrutinib, bendamustine followed by venetoclax and obinutuzumab induction and maintenance (CLL2-BAG), bendamustine followed by idelalisib and obinutuzumab induction and maintenance (CLL2-BCG), and bendamustine followed by ofatumumab and ibrutinib (CLL2-BIO). The current recommendation is to treat fit patients without significant comorbidities but with known TP53 abnormalities with ibrutinib upfront, whereas ibrutinib should be given to all fit patients without TP53 abnormalities for whom first-line chemoimmunotherapy or other targeted agents failed [8]. The optimal combination partners for these treatment approaches remain to be determined, and, as discussed, several regimens are currently in clinical development. Older or frail patients with significant comorbidities, evaluated by a comprehensive geriatric assessment, should first receive optimal supporting therapies such as vaccinations or antimicrobial prophylaxes [111]. Upon development of symptomatic disease, single-agent ibrutinib and chemoimmunotherapy combinations of CD20-antibodies and chlorambucil are reasonable treatment options [8, 111]. In addition to B-cell malignancies, ibrutinib is also approved for the treatment of steroid-resistant GvHD. A comprehensive discussion of the milestones in the development of ibrutinib in this setting, underlying mechanisms of action and current clinical trials can be found elsewhere and is beyond the scope of the current review [112].
8.2 Possible Further Indications
Ibrutinib is being explored and has shown promising therapeutic efficacy in a variety of additional indications. First, it appears that BTK inhibition might be a suitable strategy to elicit graft-versus-leukemia effects after allogeneic transplantation [113]. BTK inhibition also shows promising activity in other previously unexamined B-cell malignancies such as primary lymphomas of the central nervous system [114], whereas its single-agent efficacy has, at most, been moderate in patients with relapsed follicular lymphoma [115]. Responses have also been reported in patients with advanced solid tumors such as lung, breast, gastrointestinal and genitourinary cancers [116], both as a single agent and in combination. While off-target effects of BTK inhibition largely account for the toxicities and side effects described, it has also been proven beneficial in the restoration of cellular functions of immune cells that also express BTK or other Tec family kinase members [117]. As a result, BTK inhibitor treatment has been reported to correct CLL-induced T-cell and myeloid skewing and defects in patients treated with ibrutinib or acalabrutinib [118, 119]. Several efforts are underway to apply the immunomodulatory mode of action of BTK inhibitors to settings of autoimmune diseases and infection, which have been reviewed elsewhere [116].
9 Summary
Bruton’s tyrosine kinase inhibitors and other targeted agents have substantially altered the management, clinical course and outcomes for patients with B-cell malignancies, especially for those with previously limited treatment options due to disease characteristics or coexisting diseases. In addition, recently published phase III clinical studies highlighted that ibrutinib-based regimens are also superior to chemoimmunotherapy in previously untreated patients, especially those with CLL. However, many questions remain unanswered, including optimal combination regimens, sequencing of agents and combinations, and duration of ibrutinib-based therapy, especially with regards to MRD eradication. Moreover, a better understanding of how corrected immune functions of non-malignant cells contribute to long-term tumor control and potentially eradication will be needed. This will also necessitate ongoing revisions of the currently available prognostic models such as the GCLLSG prognostic index in CLL or the International Prognostic Index in lymphomas. Moreover, long-term and continuing treatment will pose additional challenges such as changes in the biological behavior of tumors, treatment-related mutations or adaptive mechanisms, or the development of long-term serious events such as treatment-related secondary neoplasms. Lastly, while being paradigm changing and a substantial improvement to the quality of life of patients with B-cell malignancies, the use of BTK inhibitors and other targeted agents comes at a high economic cost, and their availability might therefore be limited to certain geographical regions or healthcare settings.
References
Fischer K, Bahlo J, Fink AM, Goede V, Herling CD, Cramer P, et al. Long-term remissions after FCR chemoimmunotherapy in previously untreated patients with CLL: updated results of the CLL8 trial. Blood. 2016;127:208–15.
Thompson PA, Tam CS, O’Brien SM, Wierda WG, Stingo F, Plunkett W, et al. Fludarabine, cyclophosphamide, and rituximab treatment achieves long-term disease-free survival in IGHV-mutated chronic lymphocytic leukemia. Blood. 2016;127:303–9.
Rossi D, Terzi-di-Bergamo L, De Paoli L, Cerri M, Ghilardi G, Chiarenza A, et al. Molecular prediction of durable remission after first-line fludarabine-cyclophosphamide-rituximab in chronic lymphocytic leukemia. Blood. 2015;126:1921–4.
Eichhorst B, Fink AM, Bahlo J, Busch R, Kovacs G, Maurer C, et al. First-line chemoimmunotherapy with bendamustine and rituximab versus fludarabine, cyclophosphamide, and rituximab in patients with advanced chronic lymphocytic leukaemia (CLL10): an international, open-label, randomised, phase 3, non-inferiority trial. Lancet Oncol. 2016;17:928–42.
Goede V, Fischer K, Busch R, Engelke A, Eichhorst B, Wendtner CM, et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med. 2014;370:1101–10.
Zenz T, Gribben JG, Hallek M, Doehner H, Keating MJ, Stilgenbauer S. Risk categories and refractory CLL in the era of chemoimmunotherapy. Blood. 2012;119:4101–7.
Gribben JG. How and when I do allogeneic transplant in CLL. Blood. 2018;132:31–9.
Davids MS. How should we sequence and combine novel therapies in CLL? ASH Educ Program Book. 2017;2017:346–53.
Seda V, Mraz M. B-cell receptor signalling and its crosstalk with other pathways in normal and malignant cells. Eur J Haematol. 2015;94:193–205.
Pal Singh S, Dammeijer F, Hendriks RW. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol Cancer. 2018;17:57.
Ten Hacken E, Burger JA. Microenvironment interactions and B-cell receptor signaling in chronic lymphocytic leukemia: implications for disease pathogenesis and treatment. Biochem Biophys Acta. 2016;1863:401–13.
Weber ANR, Bittner Z, Liu X, Dang TM, Radsak MP, Brunner C. Bruton’s tyrosine kinase: an emerging key player in innate immunity. Front Immunol. 2017;8:1454.
Dubovsky JA, Beckwith KA, Natarajan G, Woyach JA, Jaglowski S, Zhong Y, et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood. 2013;122:2539–49.
Ren L, Campbell A, Fang H, Gautam S, Elavazhagan S, Fatehchand K, et al. Analysis of the effects of the Bruton’s tyrosine kinase (Btk) inhibitor ibrutinib on monocyte Fcgamma receptor (FcgammaR) function. J Biol Chem. 2016;291:3043–52.
Ping L, Ding N, Shi Y, Feng L, Li J, Liu Y, et al. The Bruton’s tyrosine kinase inhibitor ibrutinib exerts immunomodulatory effects through regulation of tumor-infiltrating macrophages. Oncotarget. 2017;8:39218–29.
Chen SS, Chang BY, Chang S, Tong T, Ham S, Sherry B, et al. BTK inhibition results in impaired CXCR4 chemokine receptor surface expression, signaling and function in chronic lymphocytic leukemia. Leukemia. 2016;30:833–43.
Andersen MA, Eriksen CT, Brieghel C, Biccler JL, Cunha-Bang CD, Helleberg M, et al. Incidence and predictors of infection among patients prior to treatment of chronic lymphocytic leukemia: a Danish nationwide cohort study. Haematologica. 2018;103(7):e300–3.
U.S. Food and Drug Administration, C.f.D.E.a.R. Imbruvica® (ibrutinib), for oral use: highlights of prescribing information (2018). https://www.imbruvica.com/docs/librariesprovider7/default-document-library/prescribing-information.pdf. Accessed 30 Oct 2018.
Advani RH, Buggy JJ, Sharman JP, Smith SM, Boyd TE, Grant B, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol. 2013;31:88–94.
Noy A, de Vos S, Thieblemont C, Martin P, Flowers CR, Morschhauser F, et al. Targeting Bruton tyrosine kinase with ibrutinib in relapsed/refractory marginal zone lymphoma. Blood. 2017;129:2224–32.
Treon SP, Tripsas CK, Meid K, Warren D, Varma G, Green R, et al. Ibrutinib in previously treated Waldenstrom’s macroglobulinemia. N Engl J Med. 2015;372:1430–40.
Dimopoulos MA, Trotman J, Tedeschi A, Matous JV, Macdonald D, Tam C, et al. Ibrutinib for patients with rituximab-refractory Waldenstrom’s macroglobulinaemia (iNNOVATE): an open-label substudy of an international, multicentre, phase 3 trial. Lancet Oncol. 2017;18:241–50.
Treon SP, Gustine J, Meid K, Yang G, Xu L, Liu X, et al. Ibrutinib monotherapy in symptomatic, treatment-naive patients with Waldenstrom macroglobulinemia. J Clin Oncol. 2018;36:2755–61.
Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369:507–16.
Rule S, Dreyling M, Goy A, Hess G, Auer R, Kahl BS, et al. Median 3.5-year follow-up of ibrutinib treatment in patients with relapsed/refractory mantle cell lymphoma: a pooled analysis. Blood. 2017;130:151.
Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369:32–42.
Stauder R, Eichhorst B, Hamaker ME, Kaplanov K, Morrison VA, Osterborg A, et al. Management of chronic lymphocytic leukemia (CLL) in the elderly: a position paper from an international Society of Geriatric Oncology (SIOG) Task Force. Ann Oncol. 2017;28:218–27.
O’Brien S, Furman RR, Coutre S, Flinn IW, Burger JA, Blum K, et al. Single-agent ibrutinib in treatment-naive and relapsed/refractory chronic lymphocytic leukemia: a 5-year experience. Blood. 2018;131:1910–9.
Farooqui MZ, Valdez J, Martyr S, Aue G, Saba N, Niemann CU, et al. Ibrutinib for previously untreated and relapsed or refractory chronic lymphocytic leukaemia with TP53 aberrations: a phase 2, single-arm trial. Lancet Oncol. 2015;16:169–76.
Ahn IE, Farooqui MZH, Tian X, Valdez J, Sun C, Soto S, et al. Depth and durability of response to ibrutinib in CLL: 5-year follow-up of a phase 2 study. Blood. 2018;131:2357–66.
O’Brien S, Jones JA, Coutre SE, Mato AR, Hillmen P, Tam C, et al. Ibrutinib for patients with relapsed or refractory chronic lymphocytic leukaemia with 17p deletion (RESONATE-17): a phase 2, open-label, multicentre study. Lancet Oncol. 2016;17:1409–18.
Byrd JC, Brown JR, O’Brien S, Barrientos JC, Kay NE, Reddy NM, et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med. 2014;371:213–23.
O’Brien S, Furman RR, Coutre SE, Sharman JP, Burger JA, Blum KA, et al. Ibrutinib as initial therapy for elderly patients with chronic lymphocytic leukaemia or small lymphocytic lymphoma: an open-label, multicentre, phase 1b/2 trial. Lancet Oncol. 2014;15:48–58.
Jones J, Mato A, Coutre S, Byrd JC, Furman RR, Hillmen P, et al. Evaluation of 230 patients with relapsed/refractory deletion 17p chronic lymphocytic leukaemia treated with ibrutinib from 3 clinical trials. Br J Haematol. 2018;182:504–12.
Dreyling M, Jurczak W, Jerkeman M, Silva RS, Rusconi C, Trneny M, et al. Ibrutinib versus temsirolimus in patients with relapsed or refractory mantle-cell lymphoma: an international, randomised, open-label, phase 3 study. Lancet. 2016;387:770–8.
Burger JA, Tedeschi A, Barr PM, Robak T, Owen C, Ghia P, et al. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N Engl J Med. 2015;373:2425–37.
Barr PM, Robak T, Owen C, Tedeschi A, Bairey O, Bartlett NL, et al. Sustained efficacy and detailed clinical follow-up of first-line ibrutinib treatment in older patients with chronic lymphocytic leukemia: extended phase 3 results from RESONATE-2. Haematologica. 2018;103:1502–10.
Woyach JA, Ruppert AS, Heerema NA, Zhao W, Booth AM, Ding W, et al. Ibrutinib regimens versus chemoimmunotherapy in older patients with untreated CLL. N Engl J Med. 2018;379:2517–28.
Burger JA, Keating MJ, Wierda WG, Hartmann E, Hoellenriegel J, Rosin NY, et al. Safety and activity of ibrutinib plus rituximab for patients with high-risk chronic lymphocytic leukaemia: a single-arm, phase 2 study. Lancet Oncol. 2014;15:1090–9.
Jain P, Keating MJ, Wierda WG, Sivina M, Thompson PA, Ferrajoli A, et al. Long-term follow-up of treatment with ibrutinib and rituximab in patients with high-risk chronic lymphocytic leukemia. Clin Cancer Res. 2017;23:2154–8.
Shanafelt TD, Wang V, Kay NE, Hanson CA, O’Brien SM, Barrientos J, et al. A randomized phase III study of ibrutinib (PCI-32765)-based therapy vs. standard fludarabine, cyclophosphamide, and rituximab (FCR) chemoimmunotherapy in untreated younger patients with chronic lymphocytic leukemia (CLL): a trial of the ECOG-ACRIN Cancer Research Group (E1912). Blood. 2018;132:LBA-4.
Dimopoulos MA, Tedeschi A, Trotman J, Garcia-Sanz R, Macdonald D, Leblond V, et al. Phase 3 trial of ibrutinib plus rituximab in Waldenstrom’s macroglobulinemia. N Engl J Med. 2018;378:2399–410.
Wang ML, Lee H, Chuang H, Wagner-Bartak N, Hagemeister F, Westin J, et al. Ibrutinib in combination with rituximab in relapsed or refractory mantle cell lymphoma: a single-centre, open-label, phase 2 trial. Lancet Oncol. 2016;17:48–56.
Jaglowski SM, Jones JA, Nagar V, Flynn JM, Andritsos LA, Maddocks KJ, et al. Safety and activity of BTK inhibitor ibrutinib combined with ofatumumab in chronic lymphocytic leukemia: a phase 1b/2 study. Blood. 2015;126:842–50.
Moreno C, Greil R, Demirkan F, Tedeschi A, Anz B, Larratt L, et al. Ibrutinib plus obinutuzumab versus chlorambucil plus obinutuzumab in first-line treatment of chronic lymphocytic leukaemia (iLLUMINATE): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2018;20:43–56.
Chanan-Khan A, Cramer P, Demirkan F, Fraser G, Silva RS, Grosicki S, et al. Ibrutinib combined with bendamustine and rituximab compared with placebo, bendamustine, and rituximab for previously treated chronic lymphocytic leukaemia or small lymphocytic lymphoma (HELIOS): a randomised, double-blind, phase 3 study. Lancet Oncol. 2016;17:200–11.
Fraser G, Cramer P, Demirkan F, Silva RS, Grosicki S, Pristupa A, et al. Updated results from the phase 3 HELIOS study of ibrutinib, bendamustine, and rituximab in relapsed chronic lymphocytic leukemia/small lymphocytic lymphoma. Leukemia. 2018. https://doi.org/10.1038/s41375-018-0276-9.
Ujjani C, Wang H, Skarbnik A, Trivedi N, Ramzi P, Khan N, Cheson BD. A phase 1 study of lenalidomide and ibrutinib in combination with rituximab in relapsed and refractory CLL. Blood Adv. 2018;2:762–8.
Jerkeman M, Eskelund CW, Hutchings M, Raty R, Wader KF, Laurell A, et al. Ibrutinib, lenalidomide, and rituximab in relapsed or refractory mantle cell lymphoma (PHILEMON): a multicentre, open-label, single-arm, phase 2 trial. Lancet Haematol. 2018;5:e109–16.
Jain N, Keating M, Thompson PA, Ferrajoli A, Burger J, Borthakur G, et al. Combined ibrutinib and venetoclax in patients with treatment-naïve high-risk chronic lymphocytic leukemia (CLL). Blood. 2018;132:696.
Hillmen P, Rawstron A, Brock K, Munoz Vincente S, Yates F, Bishop RM, et al. Ibrutinib plus venetoclax in relapsed/refractory CLL: results of the bloodwise TAP Clarity Study. Blood. 2018;132:182.
Tam CS, Anderson MA, Pott C, Agarwal R, Handunnetti S, Hicks RJ, et al. Ibrutinib plus venetoclax for the treatment of mantle-cell lymphoma. N Engl J Med. 2018;378:1211–23.
Rogers KA, Huang Y, Ruppert AS, Awan FT, Heerema NA, Hoffman C, et al. Phase 1b study of obinutuzumab, ibrutinib, and venetoclax in relapsed and refractory chronic lymphocytic leukemia. Blood. 2018;132:1568–72.
Rogers KA, Huang Y, Ruppert AS, Awan F, Hoffman C, Maddocks K, et al. Phase 2 study of combination obinutuzumab, ibrutinib, and venetoclax in treatment-naive and relapsed/refractory chronic lymphocytic leukemia. Blood. 2018;132:693.
Davids MS, Kim HT, Nicotra A, Savell A, Francoeur K, Hellman JM, et al. Umbralisib in combination with ibrutinib in patients with relapsed or refractory chronic lymphocytic leukaemia or mantle cell lymphoma: a multicentre phase 1-1b study. Lancet Haematol. 2019;6:e38–47.
Nastoupil LJ, Lunning MA, Vose JM, Schreeder MT, Siddiqi T, Flowers CR, et al. Tolerability and activity of ublituximab, umbralisib, and ibrutinib in patients with chronic lymphocytic leukaemia and non-Hodgkin lymphoma: a phase 1 dose escalation and expansion trial. Lancet Haematol. 2019;6:e100–9.
Fraietta JA, Beckwith KA, Patel PR, Ruella M, Zheng Z, Barrett DM, et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood. 2016;127:1117–27.
Herman SE, Mustafa RZ, Jones J, Wong DH, Farooqui M, Wiestner A. Treatment with ibrutinib inhibits BTK- and VLA-4-dependent adhesion of chronic lymphocytic leukemia cells in vivo. Clin Cancer Res. 2015;21:4642–51.
Wodarz D, Garg N, Komarova NL, Benjamini O, Keating MJ, Wierda WG, et al. Kinetics of CLL cells in tissues and blood during therapy with the BTK inhibitor ibrutinib. Blood. 2014;123:4132–5.
Burger JA, Li KW, Keating MJ, Sivina M, Amer AM, Garg N, et al. Leukemia cell proliferation and death in chronic lymphocytic leukemia patients on therapy with the BTK inhibitor ibrutinib. JCI Insight. 2017;2:e89904.
Woyach JA, Smucker K, Smith LL, Lozanski A, Zhong Y, Ruppert AS, et al. Prolonged lymphocytosis during ibrutinib therapy is associated with distinct molecular characteristics and does not indicate a suboptimal response to therapy. Blood. 2014;123:1810–7.
Brown JR, Moslehi J, O’Brien S, Ghia P, Hillmen P, Cymbalista F, et al. Characterization of atrial fibrillation adverse events reported in ibrutinib randomized controlled registration trials. Haematologica. 2017;102:1796–805.
Wiczer TE, Levine LB, Brumbaugh J, Coggins J, Zhao Q, Ruppert AS, et al. Cumulative incidence, risk factors, and management of atrial fibrillation in patients receiving ibrutinib. Blood Adv. 2017;1:1739–48.
Reda G, Fattizzo B, Cassin R, Mattiello V, Tonella T, Giannarelli D, et al. Predictors of atrial fibrillation in ibrutinib-treated CLL patients: a prospective study. J Hematol Oncol. 2018;11:79.
Guha A, Derbala MH, Zhao Q, Wiczer TE, Woyach JA, Byrd JC, et al. Ventricular arrhythmias following ibrutinib initiation for lymphoid malignancies. J Am Coll Cardiol. 2018;72:697–8.
Lipsky AH, Farooqui MZ, Tian X, Martyr S, Cullinane AM, Nghiem K, et al. Incidence and risk factors of bleeding-related adverse events in patients with chronic lymphocytic leukemia treated with ibrutinib. Haematologica. 2015;100:1571–8.
Wang ML, Blum KA, Martin P, Goy A, Auer R, Kahl BS, et al. Long-term follow-up of MCL patients treated with single-agent ibrutinib: updated safety and efficacy results. Blood. 2015;126:739–45.
Caron F, Leong DP, Hillis C, Fraser G, Siegal D. Current understanding of bleeding with ibrutinib use: a systematic review and meta-analysis. Blood Adv. 2017;1:772–8.
Kazianka L, Drucker C, Skrabs C, Thomas W, Melchardt T, Struve S, et al. Ristocetin-induced platelet aggregation for monitoring of bleeding tendency in CLL treated with ibrutinib. Leukemia. 2017;31:1117–22.
Varughese T, Taur Y, Cohen N, Palomba ML, Seo SK, Hohl TM, Redelman-Sidi G. Serious infections in patients receiving ibrutinib for treatment of lymphoid malignancies. Clin Infect Dis. 2018;67:687–92.
Ghez D, Calleja A, Protin C, Baron M, Ledoux MP, Damaj G, et al. Early-onset invasive aspergillosis and other fungal infections in patients treated with ibrutinib. Blood. 2018;131:1955–9.
Bercusson A, Colley T, Shah A, Warris A, Armstrong-James D. Ibrutinib blocks Btk-dependent NF-kB and NFAT responses in human macrophages during Aspergillus fumigatus phagocytosis. Blood. 2018;132:1985–8.
Rogers KA, Luay M, Zhao Q, Wiczer T, Levine L, Zeinab EB, et al. Incidence and type of opportunistic infections during ibrutinib treatment at a single academic center. Blood. 2017;130:830.
Maddocks KJ, Ruppert AS, Lozanski G, Heerema NA, Zhao W, Abruzzo L, et al. Etiology of ibrutinib therapy discontinuation and outcomes in patients with chronic lymphocytic leukemia. JAMA Oncol. 2015;1:80–7.
Sun C, Tian X, Lee YS, Gunti S, Lipsky A, Herman SE, et al. Partial reconstitution of humoral immunity and fewer infections in patients with chronic lymphocytic leukemia treated with ibrutinib. Blood. 2015;126:2213–9.
O’Brien SM, Jaglowski S, Byrd JC, Bannerji R, Blum KA, Fox CP, et al. Prognostic factors for complete response to ibrutinib in patients with chronic lymphocytic leukemia: a pooled analysis of 2 clinical trials. JAMA Oncol. 2018;4:712–6.
Thompson PA, O’Brien SM, Xiao L, Wang X, Burger JA, Jain N, et al. beta2 -microglobulin normalization within 6 months of ibrutinib-based treatment is associated with superior progression-free survival in patients with chronic lymphocytic leukemia. Cancer. 2016;122:565–73.
Barr PM, Brown JR, Hillmen P, O’Brien S, Barrientos JC, Reddy NM, et al. Impact of ibrutinib dose adherence on therapeutic efficacy in patients with previously treated CLL/SLL. Blood. 2017;129:2612–5.
Thompson PA, O’Brien SM, Wierda WG, Ferrajoli A, Stingo F, Smith SC, et al. Complex karyotype is a stronger predictor than del(17p) for an inferior outcome in relapsed or refractory chronic lymphocytic leukemia patients treated with ibrutinib-based regimens. Cancer. 2015;121:3612–21.
Miller CR, Ruppert AS, Heerema NA, Maddocks KJ, Labanowska J, Breidenbach H, et al. Near-tetraploidy is associated with Richter transformation in chronic lymphocytic leukemia patients receiving ibrutinib. Blood Adv. 2017;1:1584–8.
Woyach JA, Furman RR, Liu T-M, Ozer HG, Zapatka M, Ruppert AS, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370:2286–94.
Liu TM, Woyach JA, Zhong Y, Lozanski A, Lozanski G, Dong S, et al. Hypermorphic mutation of phospholipase C, gamma2 acquired in ibrutinib-resistant CLL confers BTK independency upon B-cell receptor activation. Blood. 2015;126:61–8.
Ahn IE, Underbayev C, Albitar A, Herman SE, Tian X, Maric I, et al. Clonal evolution leading to ibrutinib resistance in chronic lymphocytic leukemia. Blood. 2017;129:1469–79.
Woyach JA, Ruppert AS, Guinn D, Lehman A, Blachly JS, Lozanski A, et al. BTK(C481S)-mediated resistance to ibrutinib in chronic lymphocytic leukemia. J Clin Oncol. 2017;35:1437–43.
Burger JA, Landau DA, Taylor-Weiner A, Bozic I, Zhang H, Sarosiek K. Clonal evolution in patients with chronic lymphocytic leukaemia developing resistance to BTK inhibition. Nat Commun. 2016;7:11589.
Landau DA, Sun C, Rosebrock D, Herman SE, Fein J, Sivina M, et al. The evolutionary landscape of chronic lymphocytic leukemia treated with ibrutinib targeted therapy. Nat Commun. 2017;8:2185.
Scherer F, Kurtz DM, Newman AM, Stehr H, Craig AF, Esfahani MS, et al. Distinct biological subtypes and patterns of genome evolution in lymphoma revealed by circulating tumor DNA. Sci Transl Med. 2016;8:364ra155.
Dubois S, Viailly PJ, Mareschal S, Bohers E, Bertrand P, Ruminy P, et al. Next-generation sequencing in diffuse large B-cell lymphoma highlights molecular divergence and therapeutic opportunities: a LYSA study. Clin Cancer Res. 2016;22:2919–28.
Xu L, Tsakmaklis N, Yang G, Chen JG, Liu X, Demos M, et al. Acquired mutations associated with ibrutinib resistance in Waldenstrom macroglobulinemia. Blood. 2017;129:2519–25.
Paulus A, Akhtar S, Yousaf H, Manna A, Paulus SM, Bashir Y, et al. Waldenstrom macroglobulinemia cells devoid of BTK (C481S) or CXCR4 (WHIM-like) mutations acquire resistance to ibrutinib through upregulation of Bcl-2 and AKT resulting in vulnerability towards venetoclax or MK2206 treatment. Blood Cancer J. 2017;7:e565.
Zhao X, Lwin T, Silva A, Shah B, Tao J, Fang B, et al. Unification of de novo and acquired ibrutinib resistance in mantle cell lymphoma. Nat Commun. 2017;8:14920.
Lampson BL, Brown JR. Are BTK and PLCG2 mutations necessary and sufficient for ibrutinib resistance in chronic lymphocytic leukemia? Expert Rev Hematol. 2018;11:185–94.
Cheah CY, Chihara D, Romaguera JE, Fowler NH, Seymour JF, Hagemeister FB, et al. Patients with mantle cell lymphoma failing ibrutinib are unlikely to respond to salvage chemotherapy and have poor outcomes. Ann Oncol. 2015;26:1175–9.
Woyach JA. How I manage ibrutinib-refractory chronic lymphocytic leukemia. Blood. 2017;129:1270–4.
Ding W. Richter transformation in the era of novel agents. Hematology. 2018;2018:256–63.
Jones JA, Mato AR, Wierda WG, Davids MS, Choi M, Cheson BD, et al. Venetoclax for chronic lymphocytic leukaemia progressing after ibrutinib: an interim analysis of a multicentre, open-label, phase 2 trial. Lancet Oncol. 2018;19:65–75.
Mato AR, Hill BT, Lamanna N, Barr PM, Ujjani CS, Brander DM, et al. Optimal sequencing of ibrutinib, idelalisib, and venetoclax in chronic lymphocytic leukemia: results from a multicenter study of 683 patients. Ann Oncol. 2017;28:1050–6.
Turtle CJ, Hay KA, Hanafi LA, Li D, Cherian S, Chen X, et al. Durable molecular remissions in chronic lymphocytic leukemia treated with CD19-specific chimeric antigen receptor-modified t cells after failure of ibrutinib. J Clin Oncol. 2017;35:3010–20.
Robinson HR, Qi J, Cook EM, Nichols C, Dadashian EL, Underbayev C, et al. A CD19/CD3 bispecific antibody for effective immunotherapy of chronic lymphocytic leukemia in the ibrutinib era. Blood. 2018;132:521–32.
Ding W, LaPlant BR, Call TG, Parikh SA, Leis JF, He R, et al. Pembrolizumab in patients with CLL and Richter transformation or with relapsed CLL. Blood. 2017;129:3419–27.
Byrd JC, Harrington B, O’Brien S, Jones JA, Schuh A, Devereux S, et al. Acalabrutinib (ACP-196) in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374:323–32.
Wang M, Rule S, Zinzani PL, Goy A, Casasnovas O, Smith SD, et al. Acalabrutinib in relapsed or refractory mantle cell lymphoma (ACE-LY-004): a single-arm, multicentre, phase 2 trial. Lancet. 2018;391:659–67.
U.S. Food and Drug Administration, C.f.D.E.a.R. CALQUENCE® (alabrutinib) capsules, for oral use: highlights of prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/210259s000lbl.pdf. Accessed 30 Oct 2018.
Patel VK, Lamothe B, Ayres ML, Gay J, Cheung JP, Balakrishnan K, et al. Pharmacodynamics and proteomic analysis of acalabrutinib therapy: similarity of on-target effects to ibrutinib and rationale for combination therapy. Leukemia. 2018;32:920–30.
Niemann CU, Mora-Jensen HI, Dadashian EL, Krantz F, Covey T, Chen SS, et al. Combined BTK and PI3Kdelta inhibition with acalabrutinib and ACP-319 improves survival and tumor control in CLL mouse model. Clin Cancer Res. 2017;23:5814–23.
Walter HS, Rule SA, Dyer MJ, Karlin L, Jones C, Cazin B, et al. A phase 1 clinical trial of the selective BTK inhibitor ONO/GS-4059 in relapsed and refractory mature B-cell malignancies. Blood. 2016;127:411–9.
Walter HS, Jayne S, Rule SA, Cartron G, Morschhauser F, Macip S, et al. Long-term follow-up of patients with CLL treated with the selective Bruton’s tyrosine kinase inhibitor ONO/GS-4059. Blood. 2017;129:2808–10.
Tam C, Grigg AP, Opat S, Ku M, Gilbertson M, Anderson MA, et al. The BTK inhibitor, Bgb-3111, is safe, tolerable, and highly active in patients with relapsed/refractory B-cell malignancies: initial report of a phase 1 first-in-human trial. Blood. 2015;126:832.
Brown JR, Harb WA, Hill BT, Gabrilove J, Sharman JP, Schreeder MT, et al. Phase I study of single-agent CC-292, a highly selective Bruton’s tyrosine kinase inhibitor, in relapsed/refractory chronic lymphocytic leukemia. Haematologica. 2016;101:e295–8.
Cramer P, von Tresckow J, Bahlo J, Engelke A, Langerbeins P, Fink AM, et al. CLL2-BXX Phase II trials: sequential, targeted treatment for eradication of minimal residual disease in chronic lymphocytic leukemia. Future Oncol. 2018;14:499–513.
Woyach JA. What is the optimal management of older CLL patients? Best Pract Res Clin Haematol. 2018;31:83–9.
Jaglowski SM, Blazar BR. How ibrutinib, a B-cell malignancy drug, became an FDA-approved second-line therapy for steroid-resistant chronic GVHD. Blood Adv. 2018;2:2012–9.
Ryan CE, Sahaf B, Logan AC, O’Brien S, Byrd JC, Hillmen P, et al. Ibrutinib efficacy and tolerability in patients with relapsed chronic lymphocytic leukemia following allogeneic HCT. Blood. 2016;128:2899–908.
Grommes C, Pastore A, Palaskas N, Tang SS, Campos C, Schartz D, et al. Ibrutinib unmasks critical role of bruton tyrosine kinase in primary CNS lymphoma. Cancer Discov. 2017;7:1018–29.
Gopal AK, Schuster SJ, Fowler NH, Trotman J, Hess G, Hou JZ, et al. Ibrutinib as treatment for patients with relapsed/refractory follicular lymphoma: results from the open-label, multicenter, phase II DAWN study. J Clin Oncol. 2018;36:2405–12.
Campbell R, Chong G, Hawkes EA. Novel indications for Bruton’s tyrosine kinase inhibitors, beyond hematological malignancies. J Clin Med. 2018;7:E62.
Maharaj K, Sahakian E, Pinilla-Ibarz J. Emerging role of BCR signaling inhibitors in immunomodulation of chronic lymphocytic leukemia. Blood Adv. 2017;1:1867–75.
Long M, Beckwith K, Do P, Mundy BL, Gordon A, Lehman AM, et al. Ibrutinib treatment improves T cell number and function in CLL patients. J Clin Investig. 2017;127:3052–64.
Niemann CU, Herman SE, Maric I, Gomez-Rodriguez J, Biancotto A, Chang BY, et al. Disruption of in vivo chronic lymphocytic leukemia tumor-microenvironment interactions by ibrutinib-findings from an investigator-initiated phase II study. Clin Cancer Res. 2016;22:1572–82.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No sources of funding were used to prepare this manuscript.
Conflict of interest
Fabienne Lucas has no conflicts of interest that might be relevant to the contents of this manuscript. Jennifer A. Woyach receives research funding from Abbvie, Janssen, Acerta, Pharmacyclics, Loxo, Karyopharm, Morphosys and has consulted for Janssen and Pharmacyclics.
Rights and permissions
About this article

Cite this article
Lucas, F., Woyach, J.A. Inhibiting Bruton’s Tyrosine Kinase in CLL and Other B-Cell Malignancies. Targ Oncol 14, 125–138 (2019). https://doi.org/10.1007/s11523-019-00635-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-019-00635-7