
Abstract
While Post Traumatic Stress Disorder (PTSD) is associated with immune dysfunction, the underlying mechanisms remain unclear. Studies suggest a role for involvement of epigenetic mechanisms and microRNAs (miRNAs). Here, we examined genome-wide histone and DNA methylation in the peripheral blood mononuclear cells (PBMCs) in PTSD. We noted significant differences in histone H3 trimethylation at K4, K9, K27 and K36 sites in PTSD when compared to control. While overall DNA methylation level did not differ significantly between control and PTSD, the promoters of several individual genes (e.g., Interferon gamma (IFNG) and Interleukin (IL)-12B) were differentially methylated. ChIP-seq data revealed that the promoter of IFNG and TBX-21 was associated with the activation marker H3K4me3 in PTSD. The transcript levels of both IFNG and TBX-21 were higher in PTSD correlating well with the altered methylation patterns. Furthermore, PTSD patients showed increased expression of IL-12 in their PBMCs. Analysis of both histone and DNA methylation markers suggested that the expression of IL-12 was also possibly activated through epigenetic modification. Knockdown of lysine (K)-specific demethylase 5B (KDM5B), or inhibition of DNA (Cytosine-5-)-methyltransferase 1 (DNMT1) caused up-regulation of IL-12. Furthermore, the expression of these cytokines was also regulated by miRNAs. Our miRNA microarray identified many downregulated miRNAs in PTSD that are predicted to target IFNG and IL-12. Consequently, we showed that up-regulation of hsa-miR-193a-5p could decrease the expression of IL-12. Overall, the current study demonstrated that the elevated expression of pro-inflammatory cytokines in PTSD patients might be regulated by multiple epigenetic mechanisms and miRNAs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Post-traumatic stress disorder may develop following a traumatic event such as military combat. It is reported that 10–20 % of U.S. military service members suffer from PTSD after return from deployment to Iraq (Thomas et al. 2010). Even in general adult population, the prevalence rate of PTSD is about 3.5 % in the U.S. alone (Kessler et al. 2005). Symptoms related to stress usually are temporary for most people following a traumatic event. However, for PTSD patients, the traumatic event is so overwhelming that the stress symptoms may last for a very long time, which significantly affects the quality of life.
Although PTSD is mainly characterized as a psychiatric disorder, the symptoms of sustained stress not only affect nervous system but also other biological functions. Increasing evidence indicates that immune function is one of the dysregulated functions in PTSD patients. A broad spectrum of cytokine abnormalities have been reported in the blood samples from PTSD patients by us (Zhou et al. 2014) and others (Hoge et al. 2009). In general, the levels of pro-inflammatory cytokines are increased while those of anti-inflammatory cytokines are decreased (Smith et al. 2011; Sutherland et al. 2003; von Kanel et al. 2007). It has been hypothesized that dysregulation in the hypothalamic-pituitary-adrenal (HPA) axis contributes to the dysfunction of immune system in PTSD patients (Rohleder and Karl 2006). Insufficient regulation by cortisol has also been suggested as a possible reason (Gill et al. 2009). Despite these implications, it is unclear how the excessive inflammation is induced in PSTD patients. Our previous study and those of others suggest that epigenetic modifications and miRNAs play important roles in altering gene expression in the immune system of PTSD patients. For example, we have found that the level of IFNG is significantly increased in the PBMCs in PTSD patients and the increased IFNG is, at least in part, regulated by miRNAs (Zhou et al. 2014). A recent study suggests that DNA methylation may contribute to the altered expression of long non-coding RNA transcript H19 and IL-18 in PBMCs from PTSD patients by comparing their DNA methylation levels in military personnel before and after deployment (Rusiecki et al. 2013).
It is known that environmental factors can regulate gene expression through epigenetic modifications and the effect can last for a long time. Gene expression can be regulated epigenetically at the transcriptional level by DNA methylation and histone modifications and at the post-transcriptional level by miRNA and other non-coding RNA (Flintoft 2013; He and Hannon 2004; Rothbart and Strahl 2014). Among these epigenetic mechanisms, histone modification is probably the most complicated one. Histones can be phosphorylated, ubiquitinated, acetylated and methylated (Zhou et al. 2011), and methylation can be mono-, di- or tri-methylation (Greer and Shi 2012). Depending on the type and the site, these histone modifications have a very diverse regulatory function in gene expression. For example, trimethylation at lysine 4 in histone H3 (H3K4me3) in the promoter is usually associated with gene activation while trimethylation at lysine 27 (H3K27me3) is associated with gene repression (Bernstein et al. 2006; Roh et al. 2007; Wei et al. 2009). Trimethylation at lysine 9 in histone H3 (H3K9me3) is linked to gene silencing and trimethylation at lysine 36 (H3K36me3) is involved in transcription elongation (Bannister et al. 2005; Mikkelsen et al. 2007; Vakoc et al. 2005). As for DNA methylation, the modification usually occurs at 5-cytosine in the CpG region. A high degree of DNA methylation in the promoter area is believed to suppress transcription. On the other hand, DNA methylation level may be increased within the transcript body of active genes (Muers 2013). Furthermore, DNA methylation and histone methylation are interconnected. For instance, it has been shown that histone trimethylation at H3K9 and H3K27 facilitates DNA methylation because the methyltransferases catalyzing H3K9 and H3K27 methylation bind to DNA methyltransferases and facilitates their recruitment to the target loci (Lehnertz et al. 2003; Vire et al. 2006). Thus, the complex mechanism of epigenetic modifications is important for the regulation of gene expression in response to environmental stimuli including traumatic events.
Another level of gene regulation is played by miRNAs. MicroRNAs are ~22 nucleotides, non-coding RNAs which regulate gene expression post transcriptionally by inhibiting mRNA translation or degradation (Ambros 2004). Many previous reports have showed the role of miRNAs in influencing the expression of genes involved in immune function. For example, in PTSD patients, our lab has shown that IFNG level is altered and this alteration correlated with the down regulation of the hsa-miR-125a (Zhou et al. 2014).
To determine whether epigenetic modifications play a role in the altered immune function in PTSD patients and to identify potential biomarkers for PTSD diagnosis, we first compared genome-wide histone methylation and DNA methylation patterns in PBMCs from a healthy control and a PTSD patient using ChIP-seq and MeDIP-seq methods. Furthermore, we compared the DNA methylation levels of specific genes using the data available on NCBI’s GEO datasets (GSE21282) of whole genome DNA methylation from 77 controls and 23 PTSD cases (Uddin et al. 2010). Overall, we found a significant alteration in H3K4me3, H3K27me3, H3K36me3 and H3K9me3 patterns, while the overall DNA methylation did not change significantly. In addition, the expression of pro-inflammatory cytokines, IFNG and IL-12 was increased in PTSD patients and their expression correlated with their associated epigenetic markers. To determine that the altered expressions of IFNG and IL-12 were truly regulated by those epigenetic markers, we used methylation specific PCR and qRT-PCR in more PBMC samples, and the results were consistent with our ChIP-seq and MeDIP-seq data. Furthermore, knocking down H3K4me3 demethylation enzyme KDM5B, or inhibiting DNA methylation enzyme DNMT1, caused upregulation in the expression of IL-12B, suggesting that the above discussed epigenetic mechanisms could probably regulate the expression of IL-12 and IFNG. Αs another level of gene regulation, we identified altered expression of many miRNAs that are predicted to target the above mentioned genes. Consequently, we showed that miR-193a-5p, predicted to target IL-12B, was downregulated in PTSD and up-regulation of its expression in cultured cells decreased the expression of IL-12.
In summary, to show that epigenetic mechanisms can influence gene expression and with special attention to PTSD, we used ChIP-seq and MeDIP-seq data from one each control and PTSD PBMCs as a screening tool to identify potential target genes. Then, we confirmed our results in PBMCs from PTSD patients by increasing the sample size and, by performing in vitro experiments. In addition, we also showed that pro-inflammatory gene expression could be regulated at post transcriptional level by miRNAs. Our report has opened future directions to look further into understanding the mechanisms involved in the initiation and sustenance of the chronic inflammation in PTSD and developing therapeutic strategies in managing the disorder.
Materials and Methods
Patients and Cell Preparation
Informed consent was obtained from all individual participants included in the study. PTSD patients were Veterans of either the 1991 Persian Gulf war, or of the recent Iraq or Afghanistan wars, recruited from the local VA hospital, as described earlier (Zhou et al. 2014). Controls were age-matched healthy volunteers without any history of immunological disorders. Ten mL of peripheral blood was processed immediately using Ficoll-Paque (GE Healthcare, Uppsala, Sweden) centrifugation to isolate PBMCs and plasma. Viability of PBMC was determined by trypan-blue exclusion. We included 17 controls and 16 PTSD patients for the present study.
ChIP-Seq
Isolated PBMCs were treated with formaldehyde (at a final concentration of 1 %) to crosslink histone and DNA. After formaldehyde was quenched by adding glycine, cells were then pelleted and washed with cold PBS for 2 times. Chromatin was then digested with Micrococcal Nuclease. After nuclear membrane was disrupted by brief sonication, the sample was centrifuged and the supernatant was used for chromatin immunoprecipitation with Simple ChIP-enzymatic Chromatin IP Kit (Cell signaling, #9003, Danvers, MA). The ChIP antibodies, H3K4me3 (ab1012), H3K27me3 (ab6002), H3K9me3 (ab8898) and H3K36me3 (ab9050) were purchased from Abcam (Cambridge, MA). The cross link was reversed by treating the immunoprecipitated chromatin with proteinase K. DNA was then purified and quantified. The sequencing library was constructed using Illumina’s Chip Sequencing sample preparation kit (#1003473, Illumina Inc., San Diego, CA) according to the manufacturer’s instruction and sequenced by Illumina HiSeq2000 at Tufts University Genomic core facility.
MeDIP-Seq
Purified genomic DNA was treated with dsDNA Shearase (Zymo Research, Irvine, CA). DNA fragments with size from 200 to 400 bp were purified to construct sequencing library using Illumina Chip-seq library preparation kit. dsDNA was then denatured and immunoprecipitated with anti 5-methylcytosine antibody using a MeDIP kit from Diagenode (Denville, NJ). Precipitated DNA was purified and sequenced. The sample before immunoprecipitation was also sequenced as input control.
Data Analysis
Both ChIP-seq and MeDIP-seq libraries were sequenced with single-end reads of 50 bp. Raw sequencing reads were mapped to human genome build hg19 using Bowtie software by allowing two mismatches in the read (Langmead et al. 2009). The mapped reads were then filtered and only uniquely mapped reads were used for the downstream analysis. For ChIP-seq data, SICER was used for the peak calling (Blankenberg et al. 2010; Zang et al. 2009). The peak calling parameters were 200 bp window size and 600 bp gap size except for H3K4me3 in which 200 bp window size and 200 bp gap size were used. The peaks were visualized in the UCSC genome browser (http://genome.ucsc.edu/). The correlation heat map of these signals was generated using DiffBind software (Ross-Innes et al. 2012). Distribution of signal in various genomic features was calculated using CEAS software (Shin et al. 2009). Promoter region was defined as 3 kb upstream and downstream of transcription start site (TSS). For MeDIP-seq data, mapped reads were analyzed with MEDIPS software (Lienhard et al. 2014). The location of 1 kb upstream and downstream of TSS was generated using UCSC table browser. The DNA methylation signal was extracted and the correlation was analyzed. The miRNA regulation of IL-12 and IFN-γ pathway was analyzed using Ingenuity Pathway Analysis (IPA; Qiagen, Redwood city, CA). To analyze the DNA methylation levels of specific genes using the data from NCBI database, the average β values were obtained for each gene and compared between control (n = 77) and PTSD (n = 23). The β values correspond to the values obtained after performing microarray to detect the DNA methylation level at specific CpG islands.
Quantitative Real Time PCR (qRT-PCR) and Methylation Specific PCR
Complementary DNA was synthesized using miScript II RT kit (Qiagen, Valencia, CA) according to the manufacturer instructions. All the qRT-PCR reactions for the detection of gene transcripts were performed using iQ™ universal SYBR® Green supermix (Bio-Rad Inc., USA) with 25 ng of the cDNA as template. For the detection of mature miRNA(s), miScript SYBR® Green PCR kit (Qiagen, Valencia, CA) was used with 3 ng of the cDNA template. For methylation specific PCR, primers were designed employing software available online on http://www.urogene.org/cgi-bin/methprimer/methprimer.cgi (Li and Dahiya 2002). We used the sequence of the promoter region including the transcription start site (TSS) of IL-12B gene for designing the primers. The primer sequences provided by the software are as follows: Methylated (MSP) forward- AGTTGTTGGGGTAGTATATTAACGG, reverse- TATTTCAAAACCATTAAACTCTCCG, Unmethylated (USP) forward- TGTTAGTTGTTGGGGTAGTATATTAATG, reverse- ATTTCAAAACCATTAAACTCTCCAT. As per UCSC genome browser, there is one CpG island within 1Kb upstream of the TSS of IL12-B gene. The genomic location of human IL-12B gene spans from nucleotides 159314783 to 159330473 on chromosome 5. Thereafter, the DNA samples were treated using a Bisulfite conversion kit (EpiTect Bisulfite kit, Qiagen) according to the manufacturer instructions. Subsequently, PCR was performed with the bisulfite converted DNA samples. The amplified product was examined in a 2 % agarose gel.
Plasma Cytokine Assay
Plasma samples stored in −80 °C after collection, as per method described above, were used for the detection of IL-12 (p70) and 26 other cytokines by using Bioplex assay kit from Bio-Rad (Bio-Rad Inc., CA, USA) following manufacturer’s instructions and the protocol previously described by Zhou et al. 2014.
In Vitro Inhibition of DNMT1 with 5-Azacytidine (5-AZA)
THP-1 cells were cultured in complete RPMI1640 medium containing 50 ml FBS, 5 ml HEPES, 5 ml Penicillin-Streptomycin and 0.9 μl β-Mercaptoethanol every 500 ml medium. Two million cells in exponential phase were then treated with 1 μM 5-AZA in 1 ml medium for 2–4 days. Every 24 h, half of the medium was replaced with fresh medium and 5-AZA added. Total RNA was isolated from the cells after the mentioned duration of treatment and used for qRT-PCR to detect IL-12B transcripts.
Knockdown of KDM5B with siRNA
Kdm5b was knocked down in THP-1 cells by transfecting siRNA specific for it. The siRNA, with the following sequence, was procured from Integrated DNA Technologies (IDT, Iowa, USA); sense: rGrCrCrArCrCrArUrUrUrGrCrArUrGrUrGrArUdTdT and antisense: rArUrCrArCrArUrGrCrArArArUrGrGrUrGrGrCdTdT. First, 2x105 THP-1 cells were seeded in 24 well plates in 500 μl complete RPMI medium. After 24 h, 60 % of the medium was removed and the cells were transfected with siRNA encapsulated in Lipofectamine® RNAi MAX (Life Technologies, USA) at a final concentration of 5pmol following the instructions available with the transfection reagent. This was followed by addition of fresh medium equal to the volume removed before transfection. Twenty four hours after transfection, recombinant human IFNG (100 ng/ml, Biolegend, USA) and bacterial LPS (1 μg/ml, SIGMA, USA) were added to stimulate THP-1 cells to induce IL-12 expression. After stimulation for 24 hours, the cells were harvested and total RNA extracted for qRT-PCR determination of IL-12B and KDM5B transcripts.
miR-193a-5p and Anti-miR-193a-5p Transfection
Fresh PBMCs (1x106) isolated from a healthy control were transfected with either pre-miR or inhibitor (50nM final concentration) encapsulated in HiPerFect® Transfection reagent (all from Qiagen, Valencia, CA) following the manufacturer instructions. Control/mock transfection was performed with only transfection reagent but no miRNA. After transfection and making up the final volume for culture at the end of 6 h, human recombinant IFNG (100 ng/ml) and bacterial LPS (1 μg/ml) were added. The cells were then incubated for 24 h in a 5 % CO2 incubator. After incubation for the mentioned duration, total RNA was isolated and used for qRT-PCR estimation of IL-12B transcripts.
Results
Genome-Wide Histone H3 Trimethylation in PBMC
To determine whether the overall histone methylation status was altered in immune cells from PTSD patients, we isolated PBMCs from the peripheral blood and performed ChIP-seq. Four relatively well studied histone markers, H3K4me3, H3K27me3, H3K36me3 and H3K9me3 were examined. The signal intensity obtained from ChIP-seq of these markers along the 23 chromosomes is shown as a Circos plot in Fig. 1a. Overall, there were more regions in the PTSD sample associated with H3K4me3, H3K36me3 and H3K9me3 markers. However, the numbers of genomic regions associated with H3K27me3 were similar between the control and PTSD. This was further illustrated in the correlation analysis as shown in Fig. 1b. It is known that these histone markers are not evenly distributed across the genome (Mikkelsen et al. 2007; Roh et al. 2006). Their distribution patterns were also compared based on genomic features. The signal distribution profile of those 4 markers differed significantly between the control and PTSD patient (Fig. 2a–d), suggesting a shift in the location of these histone markers in the genome.

Genome-wide histone methylation in PBMCs. PBMCs from a control and a PTSD patient were isolated and histone methylation was examined by ChIP-seq as described in Methods. a) Circos plot showing histone trimethylations in the 23 chromosomes in control and PTSD. Starting from the outermost circle, the circles represent H3K4me3, H3K36me3, H3K27me3 and H3K9me3, respectively, in control and PTSD. b) The heat map of overall correlation of histone markers

Distribution of histone methylation signal among genomic features. PBMCs from control and PTSD patients were isolated as described in Fig. 1. The percentages of histone methylation signal located in the promoter region (3 kb upstream of TSS), gene body (intron and exon) and intergenic region are represented. The percentage of genomic sequence of these 3 regions is also shown. a) H3K4me3, b) H3K27me3, c) H3K36me3 and d) H3K9me3
Global DNA Methylation in PBMC
Inasmuch as DNA methylation also plays an important role in regulating gene expression, we compared global DNA methylation pattern in PBMCs from a control and a PTSD patient using MeDIP-seq. Reads that align to several positions in the genome are common in our MeDIP experiments, because of high abundance of methylation at repetitive regions. Here, we used only uniquely mapped reads. About 70-80 % of total sequence reads were uniquely mapped to the genomes. In contrast to histone methylation, the overall level and pattern of DNA methylation did not differ dramatically between the control and PTSD. The DNA methylation levels for the 23 chromosomes in both the control and PTSD patient are shown as a Circos plot in Fig. 3a. Many genes have CpG islands in their transcription start site (TSS) and DNA methylation signal is usually enriched in CpG islands. Enrichment analysis indeed showed that DNA methylation signal was highly enriched within 1 kb of TSS in both samples (Fig. 3b). Since DNA methylation in the promoter region is known to regulate gene activity, we performed signal correlation within 1 kb of TSS of all genes. The overall DNA methylation level in the promoter region correlated well between the control and PTSD (Fig. 3c), indicating that most genes had similar DNA methylation level near the TSS in these two samples. However, the signal density was significantly different for some individual genes, suggesting that the expression of those genes might be altered through DNA methylation in PBMCs from PTSD patients.

Genome-wide DNA methylation pattern in PBMCs. Genomic DNA was isolated from PBMCs of a control and a PTSD patient. DNA methylation was examined by MeDIP-seq. a) Circos plot showing DNA methylation observed in the 23 chromosomes with controls depicted in the outer circle and PTSD patient in the inner circle. The colored lines connect location of miRNAs in different chromosomes and the location of the predicted or known targets of the miRNAs. b) The relative enrichment profile of DNA methylation signal near the TSS. c) Correlation of DNA methylation signal within 1 kb of TSS between control and PTSD sample
Expression of IFNG and its Associated Histone and DNA Methylation
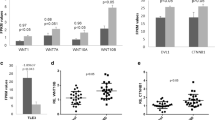
In our previous study, we showed that the protein level of IFNG was significantly increased in PBMCs from PTSD patients (Zhou et al. 2014). To determine whether the increased protein was due to the enhanced transcription, we examined the expression of IFNG and its transcription factor TBX-21 by real time PCR. The expression of both TBX-21 and IFNG was significantly increased in PTSD samples compared with the control samples (Fig. 4c), suggesting that the transcription of IFNG might be enhanced in PTSD. Examining the histone methylation from our ChIP-seq data revealed that IFNG gene in PTSD was associated with the activation marker H3K4me3 while this marker was absent in the control (Fig. 4a). Similarly, more H3K4me3 marker was found in TBX-21 gene in PTSD (Fig. 4a). In addition, we also found that the DNA methylation level near TSS of IFNG was lower in PTSD compared to that in the control (Fig. 4b). However, no significant difference in DNA methylation in TBX-21 promoter was found (data not shown).

Elevated expressions of IFNG and TBX-21 in PBMCs from PTSD patients. PBMCs from control and PTSD patients were isolated as described in Fig. 1. a) Differentially associated histone methylation marker in IFNG and TBX-21. b) DNA methylation level within the promoter region of IFNG. c, d) Relative abundance (expressed as fold change) of IFNG and TBX-21 transcripts in PBMCs from controls (n = 17) and PTSD patients (n = 16) as determined by real time PCR, respectively. Each square in the PTSD group represents one human subject. Level in the control was set as 1
Expression of IL-12 and its Associated DNA and Histone Methylation
In our MeDIP-seq data, we found that the DNA methylation in the promoter of IL-12B gene was significantly lower in PTSD than that in the control (Fig. 5a). To determine whether the differential DNA methylation level in IL-12B gene was because of individual variation or PTSD, DNA methylation specific PCR was performed using DNA isolated from PBMCs from several controls and PTSD patients. The methylated bands were much stronger than the unmethylated bands in the control samples, while the intensities of methylated and unmethylated bands in PTSD samples were similar (Fig. 5b). Furthermore, the promoter region of IL-12B gene was associated with the activation histone marker H3K4me3 in PTSD sample while a suppressive marker, H3K9me3, was found in the control sample (Fig. 5c). Both histone and DNA methylation markers suggested that the expression of this pro-inflammatory cytokine was activated through epigenetic modification. Indeed, the expression of IL-12B transcript was higher in PBMCs from PTSD samples than in control samples as determined by real time PCR (Fig. 5e). The expression of IL-12A gene, encoding the p35 subunit of IL-12, was also slightly increased in PTSD (Fig. 5d). However, no differentially associated histone marker or DNA methylation was identified in our ChIP-seq and MeDIP-seq data (data not shown), suggesting that IL-12A gene might be regulated by other mechanisms. Comparison of the DNA methylation levels in the CpG islands corresponding to IL-12B, using the NCBI’s GEO datasets, we found that the average β values of PTSD was slightly lower than that of controls (data not shown), indicating a lower DNA methylation trend in PTSD.

Elevated expression of IL-12 in PBMCs from PTSD patients. PBMCs from control and PTSD patients were isolated as described in Fig. 1. a) DNA methylation in the promoter region of IL-12B. b) Results of DNA methylation specific PCR from representative controls and PTSD patients (M: methylated DNA, U: unmethylated DNA). c) Associated histone methylation markers in IL-12B gene. d, e) Relative abundance (expressed as fold change) of IL-12A and IL-12B transcripts in PBMCs from controls (n = 17) and PTSD patients (n = 16) as determined by real time PCR, respectively. Each square in the PTSD group represents one human subject. Level in the control was set as 1
Knockdown of KDM5B or Inhibition of DNMT1 Up-Regulated the Expression IL-12B
KDM5B is a histone demethylating enzyme and is known to demethylate lysine 4 of H3K4me3 specifically. H3K4me3 type of methylation is associated with up-regulation of the target genes. After transfection with siRNA for KDM5B, the transcript level of IL-12B increased significantly when compared to control (Fig. 6c). In line with the transfection to knockdown, the transcript level of KDM5B was less in the transfected cells (Fig. 6d). DNMT1 is one of the enzymes responsible for methylation of DNA and, at low dose 5-AZA is known to inhibit this enzyme. Upon treatment with 1 μM concentration of 5-AZA, the transcripts of IL-12B was significantly higher compared to THP-1 cells that were not treated (Fig. 6b).

IL-12B expression is influenced by multiple epigenetic mechanisms and miRNAs. a) IL12 (p70) level in serum of control and PTSD subjects as analyzed by Bioplex assay. For the in vitro studies, THP-1 cells were treated with 5-AZA @1 μM concentration in one type of experiment, and transfected with siRNA for KDM5B in another, followed by quantification of IL-12B transcript by real time PCR. b) Transcript level of IL-12B after treatment with 5-AZA for 48 h. c) Transcript level of IL-12B after knockdown of KDM5B. d) Transcript level of KDM5B after it was knocked down by siRNA. e) MiRNA-gene interactive network generated in IPA for the dysregulated miRNAs and some of the select targets. Green indicates down regulated miRNAs and red is for up regulated. The solid arrows indicate direct interaction and broken for indirect. Subsequently, hsa-miR-193a-5p was up-regulated in THP-1 cells by transfection with its pre-miR as described in methods to see its effect on IL-12 expression. f) Level of hsa-miR-193a-5p in the PBMCs of PTSD detected by qRT-PCR (control 17 and PTSD 16 subjects). g) Level of IL-12B transcripts in the PBMCs from a healthy control after transfection with pre-miR-193a-5p or its inhibitor. h) IL-12 (p70) detected by ELISA in the culture supernatant after transfection of PBMCs from a healthy control with pre-miR-193a-5p. (RA relative abundance, RE relative expression)
Role of miRNA in the Expression of Pro-Inflammatory Cytokines
We reported previously that increased IFNG in PBMCs from PTSD patients was, at least in part, regulated by miRNAs (Zhou et al. 2014). In that study, we also identified many miRNAs whose expression was significantly down-regulated in PTSD samples. To assess whether those altered miRNAs were related to the increased expression of pro-inflammatory cytokines, IFNG and IL-12, we performed a miRNA-gene interaction analysis using Ingenuity Pathway Analysis (IPA; Qiagen, Redwood city, CA). As shown in Fig. 6e, several miRNAs directly target IL-12, IFNG or TBX-21. Thus, we selected hsa-miR-193a-5p as it was shown to be down regulated in our array data and also predicted to target IL-12B. Our qRT-PCR analysis for the hsa-miR-193a-5p showed that the miRNA was expressed at least 50 percent less in PTSD (Fig. 6f). Upon transfection of THP-1 cells with pre-miR-193a-5p, the transcript level of IL-12B was clearly less in mimic-transfected group (Fig. 6g). Furthermore, upon performing ELISA, with the culture supernatant for IL-12, we could not detect it in the mimic-transfected group (Fig. 6h).
Discussion
Stress is a known factor that can induce epigenetic changes and contribute to the pathogenesis of various disorders. Most evidence of epigenetic regulation in the development of PTSD-like symptoms comes from studies using animal models, and those studies usually focus on the epigenetic mechanisms in PTSD-related psychiatric effects. Very few studies have investigated the role of epigenome in immune dysfunction in PTSD patients. In this study, we first examined global histone and DNA methylation status in PBMCs. For this, we included only one sample each from the control and PTSD groups because our goal was to use it as a screening tool for identifying target genes. Significant changes in overall level and genomic regions of 4 histone markers, particularly those involved in H3K4me3, H3K36me3 and H3K9me3, suggested that the activity of histone methyltransferases and demethylases might be altered in PTSD. These results further suggested that expression of significant number of genes might be altered by histone methylation modification in PTSD. However, we are very cautious to interpret these data because individual variation may contribute to the altered histone marks greatly. Nevertheless, stress-induced global changes in histone methylation have been implicated in rodents. It has been reported that in the rat brain, acute stress causes changes in global levels of H4K9me3 and H3K27me3 with minor changes in H3K4me3, while chronic stress reduces H3K9me3 level and increases H3K4me3 level (Hunter et al. 2009). Histone deacetylases (HDACs) has been implicated in memory formation and PTSD development. Reduced level of HDAC2 has been found in PTSD patients (Sun et al. 2013). Despite the lack of the mechanistic link between PTSD and altered expression of HDAC, these observations have raised the possibility that the activity of histone modification enzymes can affect PTSD susceptibility and development. In the future, we will examine whether the activities or expression of histone methyltransferases and demethylases are altered in PBMCs with a large number of samples from healthy control and PTSD patients.
As for DNA methylation, a couple of studies have shown that the expression of some immune function genes in PBMCs from PTSD patients is regulated by DNA methylation. Using microarrays to analyze methylation status of CpG sites in cells from peripheral blood, Uddin et al.,(2010) have shown that immune function related genes are overrepresented among unmethylated genes in PTSD. Another study also showed altered global as well as gene-specific DNA methylation pattern associated with immune function (Smith et al. 2011). For example, the expression of pro-inflammatory cytokine, IL-18 is increased in PBMCs from PTSD patients, and this increased expression correlates with a decrease in DNA methylation in the gene (Rusiecki et al. 2013). We found that the promoter region in many genes in PBMCs of PTSD has different DNA methylation levels when compared to control, even though the overall methylation profile does not change significantly. Thus, our data suggest that the expression of many of the genes involved in the pathogenesis of PTSD and the inflammation associated with it could be modified because of altered DNA methylation in specific genes.
IL-12 is a pro-inflammatory gene whose role in PTSD-related immune dysfunction has not been reported. Functionally, IL-12 (p70) has IL-12A (p35) and IL-12B (p40) subunits, which are combined as a heterodimer. Our screening for altered gene expression indicated that IL-12B expression is probably deregulated in PTSD. Correlating with this observation, qRT-PCR analysis with bigger sample size indeed showed that IL-12B gene transcript was more in PTSD. Our histone methylation data showed a higher level of H3K4me3 around IL-12B promoter in PTSD, which is indicative of a higher transcription of the gene. Thus, to confirm that IL-12B transcription is influenced by H3K4me3 methylation, we knocked down KDM5B by using siRNA. KDM5B is the enzyme responsible for demethylation of H3K4me3 (Klein et al. 2014). We hypothesized that knocking down KDM5B will result in decreased demethylation of H3K4me3 leading to a higher transcription of IL-12B gene. Indeed, we saw higher transcript level of IL-12B after siRNA knockdown of KDM5B. Moreover, upon confirmation by qRT-PCR, the level of KDM5B was less in the siRNA transfected cells compared to vehicle indicating that the knockdown was effective. These data indicated that IL-12B gene expression is influenced by H3K4me3 methylation.
Additionally, our screening result indicated that DNA methylation was lower in the promoter of IL-12B in PTSD. Thus, we performed methylation specific PCR with primers encompassing the CpG island around the IL-12B promoter after including more samples. We found that the methylated bands were much stronger than the unmethylated bands in the control samples, while the intensities of methylated and unmethylated bands in PTSD samples were similar. DNA methylation around promoter region is associated with lower transcription of the corresponding gene (Muers 2013). Thus, our results indicated that the promoter of IL-12B gene was heavily methylated in the control individuals and the expression of this gene might be suppressed. On the other hand, DNA methylation on IL-12B promoter was lower in PTSD and thereby the methylation-mediated suppression might be lifted in PTSD. To confirm that IL-12B gene expression is influenced by DNA methylation as well, we set to alter DNA methylation in THP-1 cells. We treated the cells with 5-Azacytidine (5-AZA) to inhibit DNA methyltransferase. 5-AZA is believed to inhibit DNA methyltransferase at a lower concentration (Christman 2002). Our treatment results showed that IL-12B transcript was significantly more in the treated group compared to vehicle controls. This observation indicated that lower DNA methylation can enhance the expression of IL-12B and thus could possibly be another mechanism that leads to its altered expression in PTSD.
IFNG is a hallmark cytokine produced by Th1 cells and its expression is elevated in PBMCs from PTSD patients as shown in our previous study (Zhou et al. 2014). IL-12 is known to induce IFNG expression (Puddu et al. 1997) via the JAK-STAT signaling pathway (Schroder et al. 2004). In this study, we found that both histone marks and DNA methylation associated with IL-12B and IFNG are altered, consistent with the fact that their expression is increased in PBMCs from PTSD patients. Based on this, we hypothesize that activation of IFNG producing cells in PTSD is probably through the IL-12 signaling which involves the JAK-STAT pathway. Interestingly, our other data (not published) on JAK and STAT molecules also show that they are up regulated in PTSD compared to controls. Moreover, TBX-21, the transcription factor that binds to IFNG promoter (Djuretic et al. 2007; Collier et al. 2014) is also up-regulated in PTSD as per our qRT-PCR analysis. The H3K4me3 marker on TBX-21 was found to be more prominent in the PTSD sample as evidenced in Fig. 4a. This result suggests that the observed up-regulation of the TBX-21 gene in PTSD could be because of a higher H3K4me3 methylation.
Post-transcriptional regulation of gene expression by miRNAs is another layer of gene regulation (Bartel 2004). For example, we have already shown the relationship between dysregulated IFNG in PTSD and altered expression of miRNAs that target it (Zhou et al. 2014). In our ongoing study, employing microarray, we have identified that the expression of many miRNAs is down regulated in PBMCs from PTSD patients (data not shown). Many of the downregulated miRNAs are predicted to directly target IL-12, as shown in Fig. 6e, suggesting that the expression of the pro-inflammatory cytokine will be further increased. Thus, we sought to further study the effect of hsa-miR-193a-5p in context of IL-12B expression. We transfected pre-miR-193a-5p into PBMCs collected from healthy donor. Our results showed that transfection with miR-193a-5p lowered the level of both IL-12B transcripts and IL-12 p70. These data clearly indicated that IL-12B could be a target of hsa-miR-193a-5p and altered expression of the miRNA can influence the expression of IL-12B at the post transcriptional level. However, we understand that further study has to be performed to prove that hsa-miR-193a-5p directly interacts with IL-12B.
In summary, the current study demonstrates that the pro-inflammatory cytokine IL-12, is up-regulated in the PBMCs of PTSD, thereby suggesting a role for IL-12 in the increase in IFNG seen in these patients. Also, we provide evidence that multiple epigenetic mechanisms and miRNAs can influence the expression of pro-inflammatory genes. As a proof, we demonstrate that the expression of IL-12 and IFNG could be influenced by multiple epigenetic mechanisms (histone methylation, DNA methylation) and by miRNAs. Nonetheless, we understand that a bigger sample size and cell specific study is needed to gather more information on the genome-wide effects of PTSD on these epigenetic markers and the expression of miRNAs. A recent large study showed that men and women with PTSD had a higher incidence of infectious disease, endocrine-metabolic, neurologic, digestive, autoimmune, and numerous other disorders (Frayne et al. 2011). It is interesting to note that inflammation serves as the underlying cause of most of such clinical disorders. Thus, our studies aimed at understanding the role of epigenome and miRNA in triggering heightened inflammation in PTSD may lead to development of novel strategies to prevent and treat PTSD but also several of the PTSD-associated clincial disorders.
References
Ambros V (2004) The functions of animal microRNAs. Nature 431(7006):350–355. doi:10.1038/nature02871
Bannister AJ, Schneider R, Myers FA, Thorne AW, Crane-Robinson C, Kouzarides T (2005) Spatial distribution of di- and tri-methyl lysine 36 of histone H3 at active genes. J Biol Chem 280:17732–17736. doi:10.1074/jbc.M500796200
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116(2):281–297. doi:10.1016/S0092-8674(04)00045-5
Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, Jaenisch R, Wagschal A, Feil R, Schreiber SL, Lander ES (2006) A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125:315–326. doi:10.1016/j.cell.2006.02.041
Blankenberg D, Von Kuster G, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A, Taylor J (2010) Galaxy: a web-based genome analysis tool for experimentalists. Curr Protoc Mol Biol Chapter 19: Unit 19 10 11–21
Christman JK (2002) 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene 21(35):5483–5495. doi:10.1038/sj.onc.1205699
Collier SP, Henderson MA, Tossberg JT, Aune TM (2014) Regulation of the Th1 genomic locus from Ifng through Tmevpg1 by T-bet. J Immunol 193(8):3959–3965. doi:10.4049/jimmunol.1401099
Djuretic IM, Levanon D, Negreanu V, Groner Y, Rao A, Ansel KM (2007) Transcription factors T-bet and Runx3 cooperate to activate Ifng and silence Il4 in T helper type 1 cells. Nat Immunol 8(2):145–153. doi:10.1038/ni1424
Flintoft L (2013) Non-coding RNA: structure and function for lncRNAs. Nat Rev Genet 14:598. doi:10.1038/nrg3561
Frayne SM, Chiu VY, Iqbal S, Berg EA, Laungani KJ, Cronkite RC, Pavao J, Kimerling R (2011) Medical care needs of returning veterans with PTSD: their other burden. J Gen Intern Med 26(1):33–39. doi:10.1007/s11606-010-1497-4
Gill JM, Saligan L, Woods S, Page G (2009) PTSD is associated with an excess of inflammatory immune activities. Perspect Psychiatr Care 45:262–277. doi:10.1111/j.1744-6163.2009.00229.x
Greer EL, Shi Y (2012) Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet 13:343–357. doi:10.1038/nrg3173
He L, Hannon GJ (2004) MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet 5:522–531. doi:10.1038/nrg1379
Hoge EA, Brandstetter K, Moshier S, Pollack MH, Wong KK, Simon NM (2009) Broad spectrum of cytokine abnormalities in panic disorder and posttraumatic stress disorder. Depress Anxiet 26:447–455. doi:10.1002/da.20564
Hunter RG, McCarthy KJ, Milne TA, Pfaff DW, McEwen BS (2009) Regulation of hippocampal H3 histone methylation by acute and chronic stress. Proc Natl Acad Sci U S A 106:20912–20917. doi:10.1073/pnas.0911143106
Kessler RC, Chiu WT, Demler O, Merikangas KR, Walters EE (2005) Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry 62:617–627. doi:10.1001/archpsyc.62.6.617
Klein BJ, Piao L, Xi Y, Rincon-Arano H, Rothbart SB, Peng D, Wen H, Larson C, Zhang X, Zheng X, Cortazar MA, Peña PV, Mangan A, Bentley DL, Strahl BD, Groudine M, Li W, Shi X, Kutateladze TG (2014) The histone-H3K4-specific demethylase KDM5B binds to its substrate and product through distinct PHD fingers. Cell Rep 6(2):325–335. doi:10.1016/j.celrep.2013.12.021
Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10(3):R25. doi:10.1186/gb-2009-10-3-r25
Lehnertz B, Ueda Y, Derijck AA, Braunschweig U, Perez-Burgos L, Kubicek S, Chen T, Li E, Jenuwein T, Peters AH (2003) Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr Biol 13:1192–1200. doi:10.1016/S0960-9822(03)00432-9
Li LC, Dahiya R (2002) MethPrimer: designing primers for methylation PCRs. Bioinformatics 18(11):1427–1431. doi:10.1093/bioinformatics/18.11.1427
Lienhard M, Grimm C, Morkel M, Herwig R, Chavez L (2014) MEDIPS: genome-wide differential coverage analysis of sequencing data derived from DNA enrichment experiments. Bioinformatics 30:284–286. doi:10.1093/bioinformatics/btt650
Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, Lee W, Mendenhall E, O’Donovan A, Presser A, Russ C, Xie X, Meissner A, Wernig M, Jaenisch R, Nusbaum C, Lander ES, Bernstein BE (2007) Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448:553–560. doi:10.1038/nature06008
Muers M (2013) Gene expression: disentangling DNA methylation. Nat Rev Genet 14:519. doi:10.1038/nrg3535
Puddu P, Fantuzzi L, Borghi P, Varano B, Rainaldi G, Guillemard E, Malorni W, Nicaise P, Wolf SF, Belardelli F, Gessani S (1997) IL-12 induces IFN-gamma expression and secretion in mouse peritoneal macrophages. J Immunol 159:3490–3497
Roh TY, Cuddapah S, Cui K, Zhao K (2006) The genomic landscape of histone modifications in human T cells. Proc Natl Acad Sci U S A 103:15782–15787. doi:10.1073/pnas.0607617103
Roh TY, Wei G, Farrell CM, Zhao K (2007) Genome-wide prediction of conserved and nonconserved enhancers by histone acetylation patterns. Genome Res 17:74–81. doi:10.1101/gr.5767907
Rohleder N, Karl A (2006) Role of endocrine and inflammatory alterations in comorbid somatic diseases of post-traumatic stress disorder. Minerva Endocrinol 31:273–288
Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, Ali S, Chin SF, Palmieri C, Caldas C, Carroll JS (2012) Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481:389–393. doi:10.1038/nature10730
Rothbart SB, Strahl BD (2014) Interpreting the language of histone and DNA modifications. Biochim Biophys Acta 1839(8):627–643. doi:10.1016/j.bbagrm.2014.03.001
Rusiecki JA, Byrne C, Galdzicki Z, Srikantan V, Chen L, Poulin M, Yan L, Baccarelli A (2013) PTSD and DNA methylation in select immune function gene promoter regions: a repeated measures case–control study of U.S. Military Service Members. Front Psychiatr 4:56. doi:10.3389/fpsyt.2013.00056
Schroder K, Hertzog PJ, Ravasi T, Hume DA (2004) Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol 75(2):163–189. doi:10.1189/jlb.0603252
Shin H, Liu T, Manrai AK, Liu XS (2009) CEAS: cis-regulatory element annotation system. Bioinformatics 25:2605–2606. doi:10.1093/bioinformatics/btp479
Smith AK, Conneely KN, Kilaru V, Mercer KB, Weiss TE, Bradley B, Tang Y, Gillespie CF, Cubells JF, Ressler KJ (2011) Differential immune system DNA methylation and cytokine regulation in post-traumatic stress disorder. Am J Med Genet B Neuropsychiatr Genet 156B:700–708. doi:10.1002/ajmg.b.31212
Sun H, Kennedy PJ, Nestler EJ (2013) Epigenetics of the depressed brain: role of histone acetylation and methylation. Neuropsychopharmacology 38:124–137. doi:10.1038/npp.2012.73
Sutherland AG, Alexander DA, Hutchison JD (2003) Disturbance of pro-inflammatory cytokines in post-traumatic psychopathology. Cytokine 24:219–225. doi:10.1016/j.cyto.2003.09.004
Thomas JL, Wilk JE, Riviere LA, McGurk D, Castro CA, Hoge CW (2010) Prevalence of mental health problems and functional impairment among active component and National Guard soldiers 3 and 12 months following combat in Iraq. Arch Gen Psychiatry 67:614–623. doi:10.1001/archgenpsychiatry.2010.54
Uddin M, Aiello AE, Wildman DE, Koenen KC, Pawelec G, de Los Santos R, Goldmann E, Galea S (2010) Epigenetic and immune function profiles associated with posttraumatic stress disorder. Proc Natl Acad Sci U S A 107:9470–9475. doi:10.1073/pnas.0910794107
Vakoc CR, Mandat SA, Olenchock BA, Blobel GA (2005) Histone H3 lysine 9 methylation and HP1gamma are associated with transcription elongation through mammalian chromatin. Mol Cell 19:381–391. doi:10.1016/j.molcel.2005.06.011
Vire E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, Morey L, Van Eynde A, Bernard D, Vanderwinden JM, Bollen M, Esteller M, Di Croce L, de Launoit Y, Fuks F (2006) The Polycomb group protein EZH2 directly controls DNA methylation. Nature 439:871–874. doi:10.1038/nature04431
von Kanel R, Hepp U, Kraemer B, Traber R, Keel M, Mica L, Schnyder U (2007) Evidence for low-grade systemic proinflammatory activity in patients with posttraumatic stress disorder. J Psychiatr Res 41:744–752. doi:10.1016/j.jpsychires.2006.06.009
Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z, Cui K, Kanno Y, Roh TY, Watford WT, Schones DE, Peng W, Sun HW, Paul WE, O’Shea JJ, Zhao K (2009) Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity 30:155–167. doi:10.1016/j.immuni.2008.12.009
Zang C, Schones DE, Zeng C, Cui K, Zhao K, Peng W (2009) A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics 25:1952–1958. doi:10.1093/bioinformatics/btp340
Zhou VW, Goren A, Bernstein BE (2011) Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet 12:7–18. doi:10.1038/nrg2905
Zhou J, Nagarkatti P, Zhong Y, Ginsberg JP, Singh NP, Zhang J, Nagarkatti M (2014) Dysregulation in microRNA expression is associated with alterations in immune functions in combat veterans with post-traumatic stress disorder. PLoS One 9:e94075. doi:10.1371/journal.pone.0094075. eCollection 2014
Acknowledgments
This study is supported in part by National Institutes of Health grants P01AT003961, R01AT006888, R01ES019313, R01MH094755, and P20GM103641 as well as by Veterans Affairs Merit Award BX001357 to PN and MN.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical Approval
All procedures performed in studies involving human participants were approved by the Institutional Review Board.
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Marpe Bam and Xiaoming Yang contributed equally to this work.
Rights and permissions
About this article

Cite this article
Bam, M., Yang, X., Zhou, J. et al. Evidence for Epigenetic Regulation of Pro-Inflammatory Cytokines, Interleukin-12 and Interferon Gamma, in Peripheral Blood Mononuclear Cells from PTSD Patients. J Neuroimmune Pharmacol 11, 168–181 (2016). https://doi.org/10.1007/s11481-015-9643-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11481-015-9643-8