
Abstract
Purpose
In the present study, a method for the detection of 25 psychoactive substances in cerumen was developed and validated. This method targets opiates, cocaine, antidepressants, benzodiazepines, antipsychotics and antiparkinsons.
Methods
Analysis was performed on a SCIEX Triple Quad 6500+ system after liquid–liquid extraction. Methanol with 1% acetic acid was chosen as the extraction solvent. After the addition of the solvent, samples were vortexed, sonicated, centrifuged and directly injected into the liquid chromatography–tandem mass spectrometry system.
Results
The method was found to be selective and sensitive (limit of detection: 0.017 ng–0.33 ng/mg), the assay was linear for all analytes with linear regression coefficient ranging 0.9911–1.00. The values for intra-assay precision was between 4.34 and 14.6% and inter-assay precision between 5.81 and 17.7%, with accuracy within the acceptable criteria for all analytes. All analytes in cerumen specimens were stable for 48 h at 4 °C and 72 h at − 20 °C, whilst no significant matrix effect or carryover was observed. Applicability was proven by analyzing cerumen samples from 25 deceased with a history of drug abuse. All analytes were detected in real samples, thus confirming the sensitivity of the developed method.
Conclusions
According to our knowledge, it is the first time that a method for the simultaneous detection of 25 psychoactive drugs in cerumen was developed, fully validated and finally applied to 25 postmortem samples.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In terms of medico-legal death investigations, apart from common specimens such as blood and urine, alternative samples like tissues, hair, oral fluid or nails are collected at autopsy for toxicological testing, mainly in order to conclude on the use of drugs over extended time periods.
Since, urine analysis may cover a limited time range of several days, hair and nails analysis are routinely performed to monitor drug abuse over a period of several months [1, 2]. The major pitfalls of hair analysis are hair coloration, cosmetic procedures such as bleaching or heat treatment of hair may remove analytes prior to sample collection [3, 4], and also decontamination of hair via deposition from a contaminated atmosphere, whilst the main disadvantages of nail analysis are external decontamination and the use of nail polish and acetone that might alter substance incorporation into nails [5].
A biological specimen that has not yet been used extensively as an alternative biological matrix for drug detection is cerumen, or commonly referred to as “earwax”: a keratin-based mixture of squalene, cholesterol and wax esters, triacylglyceroles, ceramides and cholesterol sulfate, secreted by sebaceous and ceruminal glands that are located in the cartilaginous outer third of the external auditory canal [6]. It contains a high percentage of keratin as a result of epithelial migration from the tympanic membrane [7] and for this reason it is considered as a keratinized matrix as well as hair and nail [8,9,10].
Previous studies showed incorporation of drugs and drugs of abuse in cerumen [11, 12]. The incorporation mechanism of drugs is probably the same like hair and nails, since it is a keratinized matrix: drugs through blood circulation incorporate to keratin fibers [13].
Compared to blood and urine, cerumen seems to have a longer window of detection (weeks to months) [12, 14]. Cerumen collection is non-invasive, easy to perform and non-medical trained personnel can perform the sample collection. The extent of atmosphere decontamination is smaller than that of hair or nails since the ear canal is more protected from the external environment than the skin. Additionally, cerumen can be transported and stored at room temperature similarly like hair and nails.
Shokry [11] et al. managed to develop a method for the detection of neuropsychotic drugs in cerumen, whilst Meier et al. [12] developed a qualitative method for the detection of drugs of abuse in cerumen. Furthermore, Shokry et al. used cerumen to evaluate tobacco exposure in humans, by measuring the concentrations of nicotine and cotinine in cerumen and distinguishing subjects between non-smokers, passive smokers, and active smokers [15]. In another study, Meier et al. investigated the cerumen levels of 4-fluoroamphetamine after 1 and 12 h of monitored intake in 12 subjects with a liquid chromatography–tandem mass spectrometry (LC–MS/MS) technique [14]. Finally, Nicolaou et al. sampled cerumen from 4 cannabis users and developed an ultra-high-performance liquid chromatography (UHPLC)-MS/MS method for detecting and quantifying cannabinoids [16]. In this work, for the first time, we describe the development and validation of a new quantitative method for the simultaneously analysis of 25 drugs and drugs of abuse in cerumen, its application to 24 real postmortem samples of drug users and the correlation of the results with those of blood and urine. The list of drugs under investigation included the most common drugs of abuse: opiates, cocaine and benzodiazepines. The list also included widely prescribed pharmaceuticals (antipsychotics, antidepressants, antiepileptics and antiparkinsons) often abused by addicted people. LC–MS/MS was used as analytical technique for the detection of all the biological specimens used in this work, after a simple liquid–liquid extraction (LLE) with methanol. To the best knowledge of the authors there are no methods in the literature reporting the simultaneous detection and quantitation of 25 drugs of abuse in cerumen.
Materials and methods
Chemicals and reagents
Methanol and acetonitrile LC–MS grade were purchased from Fisher Scientific International Inc. (Hampton, NH, USA) and formic and acetic acid (> 98%) were purchased from Riedel-de Haën1 (Sigma Aldrich, Steinheim, Germany). HPLC grade water was obtained from a Milli-Q purification system (Millipore, Bedford, MA, USA). Compounds of the highest available purity were used as standards and internal standard were supplied by Lipomed (Arlesheim, Switzerland), Cerilliant (Texas, USA) and LGC (Luckenwalde, Germany). 6- Acetyl-morphine-D3 and nordiazepam-D5, were used as internal standard (IS) in a concentration of 25 ng/mg and 5 ng/mg cerumen, respectively.
Equipment and conditions
LC separation was performed on an ExionLC™ system (AB Sciex, Toronto, Canada) using an Acquity BEH C18 column (150 × 2.1 mm i.d., 1.7 μm; Waters Corporation, Millford, MA) protected by an Acquity BEH C18 VanGuard pre-column (5 mm × 2.1 mm i.d., 1.7 μm; Waters). Mobile phases were formic acid in water (0.1%) (mobile phase A) and formic acid in acetonitrile (mobile phase B). The injection volume was 5 µL, column temperature was set at 50 °C and the LC runtime was 12.5 min. The flow rate was maintained at 0.5 mL/min. Separation was accomplished employing 5% phase B in the first 0.2 min of the run, followed by a linear gradient from 5 to 25% phase B over the following 2.8 min. Mobile phase B reached 100% at 7.5 min of the analysis and remained in this condition for 1.5 min, then returned to the initial condition from 9.5 min to 12.5 min with phase B maintained to 5% to equilibrate the column. The eluent from the column was directed to the mass spectrometer with splitless mode. A SCIEX Triple Quad 6500+ system (AB Sciex) was used, the ionization source was operated in electrospray ionization (ESI) mode in positive polarity. For each compound, two multiple reaction monitoring (MRM) transitions were selected, one as quantifier and one qualifier for confirmative identification and each sample was injected in triplicate. Ion source parameters were optimized for the lower abundance compounds (curtain gas: 30 psi; ion source gas 1: 60 psi; ion source gas 2: 30 psi; temperature: 550 °C; collision gas (collision-induced dissociation): low; interface heater: on; needle voltage: 3500 V). The parameters and the selected transitions are summarized in Supplementary Table 1. System control and data acquisition were performed with Analyst software (AB SCIEX) including the ‘‘Explore’’ option (for chromatographic and spectral interpretation) and the ‘‘Quantitate’’ option (for quantitative information generation). Calibration curves were constructed with the Analyst Quantitation program.
Specimens
Twenty-five blood, urine and earwax samples were collected from deceased with a history of drug abuse. The samples were collected in autopsy, 14–20 h post mortem. Samples were stored at − 20 °C until analysis except for cerumen samples which were stored in a dry and dark place at room temperature until the day of analysis. Femoral vein serum was analyzed for recent drug use (screening and quantification, respectively) and urine to detect past drug use. Initially, blood and urine compound screening was performed using a gas chromatography–mass spectrometry (GC-MS) procedure [17]. Subsequently, quantitative analysis in blood samples were performed with a previously described targeted ultra-performance liquid chromatography (UPLC)–MS/MS method, [18] whilst urine samples were analyzed with a fast “dilute and shoot” targeted LC–MS/MS method according to Cao et al. [19].
Sample preparation
Sampling of cerumen was performed using cotton swabs 75 mm length, in both ears prior to autopsy to avoid contamination with blood. The cotton swabs with cerumen were accurately weighted before and after collection and stored until analysis. In the day of analysis, 600 μL of methanol with 1% acetic acid was added, samples were then vortexed for 10 min and sonicated for 30 min. Subsequently, samples were centrifuged at 21,206g for 5 min.
Drug-free weighted cerumen samples from deceased with no history of drug abuse were spiked with various concentrations of the investigated drugs in order to obtain a six point calibration curve and three calibrators, used for validation purposes. Method was designed for 5 mg of cerumen and concentration was corrected using the exact weight of each sample. Twenty μL of each working solution was added to cerumen sample.
Working solutions, calibration curve, and quality control samples
The stock and standard solutions were stored at − 20 °C. Standard solutions were diluted with methanol to obtain working solutions. Calibration points and quality control (QC) samples were prepared by mixing the appropriate working solution with blank whole blood, urine, and cerumen samples.
Calibration curves for blood and urine were constructed in duplicates over the concentration range equivalent to 5.00–1000 ng/mL for methadone, 2-ethylidene-1,5-dimethyl-3,3-diphenylpyrrolidine (EDDP), 6-acetyl-morphine, codeine, morphine, diazepam, nordiazepam, bromazepam, lorazepam, alprazolam, oxazepam, temazepam, 7-Amino-flunitrazepam (7-AF). For cocaine, benzoyl ecgonine (BZE) and ecgonine methyl ester (EME): 10.0–2000 ng/mL, for mirtazapine, citalopram, sertraline, amitriptyline, quetiapine and venlafaxine: 1.00–500 ng/mL, for haloperidol: 0.100–50.0 ng/mL, for olanzapine and biperiden: 0.20–100 ng/mL. The calibrators, used for validation purposes at three different concentrations (low, medium, high) were prepared in the same way at concentrations equivalent to 5.00 (LQC), 250 (MQC) and 750 (HQC) ng/mL, for methadone, EDDP, 6-acetyl-morphine, codeine, morphine, diazepam, nordiazepam, bromazepam, lorazepam, alprazolam, oxazepam, temazepam, 7-Amino-flunitrazepam, 10.0 (LQC), 400 (MQC), 1500 ng/mL (HQC) for cocaine, benzoyl ecgonine and ecgonine methyl ester, 1.00 (LQC), 50 (MQC) and 350 (HQC) for mirtazapine, citalopram, sertraline, amitriptyline, quetiapine and venlafaxine, 0.10 (LQC), 5.00 (MQC) and 37.5 (HQC) ng/mL for haloperidol, 0.20 (LQC), 10.0 (MQC) and 75.0 ng/mL (HQC) for olanzapine and biperiden.
For cerumen, the concentration range and the quality controls used for the validation are summarized in Table 1.
Samples with concentration over the highest point of the calibration curve, were diluted with drug-free blood or urine to fit the linear curve. For cerumen samples out of the linear curve, samples were diluted with additional volume of the extraction solvent. Dilution integrity test was performed and the results were within the acceptable range (accuracy and precision within ± 15%).
Method validation
Evaluation of method performance including limit of detection (LOD), lower limit of quantification (LLOQ), linearity, accuracy, precision, extraction recovery (RE), carryover, ion suppression (matrix effect, ME) and stability was estimated with respect to international guidelines [20,21,22].
Regression lines were assessed based on peak area ratios of the analytes to that of the internal standards and linear regression was carried out to determine the mean coefficients (R2) of the calibration curves. The LLOQ is the lowest amount of an analyte in a sample that can be quantitatively determined with suitable precision and accuracy (bias) (coefficient of variation < 20%), for LLOQ, S/N is usually required to be equal to or greater than 10 [19]. In this method, limit of quantitation (LOQ) was used as the lowest calibration point. LOD is considered the lowest concentration that gives a reproducible instrument response with S/N ratio > 3.
For the assessment of intra- and inter-day precision, six replicates of QC samples with the 25 compounds of interest at low, middle and high concentration level were analyzed. Intra-day precision was estimated within the same day, while inter-day over a period of 5 consecutive days. Accuracy was also performed in three different concentrations (LQC, MQC, HQC) and was expressed as percentage of the true value.
Potential interferences of cerumen matrix or secondary analytes were studied in selectivity experiments with cerumen drug-free samples (n = 6), according to Matuszewski et al. ME and RE were determined at low, medium and high concentration using six different blank cerumen samples [20].
To study carryover effect, a blank sample was injected after the analysis of a cerumen sample spiked at the highest concentration of the calibration curve.
Finally, stability of all compounds in extracted quality control samples was assessed at low and high concentration level. Short-term stability was evaluated for sample extracts kept in the autosampler for up to 48 h, as well as for samples extracts kept in the freezer for 72 h.
Results
Initially, sample extraction was optimized by adjusting the extraction solvent, volume, and time in sonication. Methanol with 1% acetic acid gave satisfactory results and yielded better recoveries for the selected analytes of this method. Sample extraction was optimized by examining various solvents or combinations, including methanol, ethyl acetate, acetonitrile, and/or water. We used recovery studies in order to determine the type of solvent that significantly improved the extraction efficiency of all analytes. The acidified methanol provided the best extraction efficiency, sharper chromatographic peaks and was selected as the extraction solvent for this study. All the other solvents were excluded because they resulted in lower extraction recoveries or higher background noise. The optimum extraction time and volume of extraction solvent were found to be 30 min and 0.6 mL, respectively.
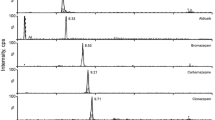
During chromatographic method optimization, methanol and acetonitrile were tested as organic mobile phase (B). Acetonitrile was chosen because it improved the chromatographic separation of the analytes and also avoided interferences caused by the matrix (Fig. 1).


Extracted ion chromatograms of fortified samples at the concentration of their LOQ. a 6-MAM, codeine, morphine; b cocaine, EME, BZE; c 7-AF, alprazolam, diazepam, nordiazepam; d bromazepam, lorazepam; e methadone, EDDP; f olanzapine, mirtazapine, quetiapine, sertraline; g venlafaxine, citalopram, haloperidol, biperiden, amitriptyline
MS conditions for cocaine and methadone were adjusted differently from the other analytes, due to their great sensibility at ESI. For these analytes, were adopted the less abundant ions (m/z 77 instead of 182, and 105 instead of 265), which allowed quantification and good linearity results.
Calibration curves were generated by plotting the peak area versus the spiked analyte concentrations. The devised method was found to be linear over the dynamic ranges for all analytes with R2 values > 0.99 (Table 1).
LOQ was found to be between 0.05 and 1.00 ng/mg depending on the analyte; LOD was between 0.017 and 0.33 ng/mg. Analytical results of the method are summarized in Table 1.
Precision and accuracy of the present method were determined by analyzing quality control samples at three different concentration levels (low, medium and high) for individual analytes. A summary of the accuracy (expressed as %) and precision (expressed as % relative standard deviation) data of the individual quality control samples for the investigated drugs is shown in Table 2. The method showed good precision as shown in Table 2 and results suggest that accuracy values fell within the acceptable criteria ranging between 80.0 and 119.8%.
Matrix effects were evaluated by comparison of target peak areas in six blank samples from different sources fortified with analytes after extraction (at low, medium and high QC levels) with the average target peak areas of a set of neat standards. The results showed that matrix did not significantly affect signal intensity. The percentage recovery of the analytes assessed by the ratio of responses of spiked cerumen prior and post extraction at three concentration levels (LQC, MQC, HQC) ranged from 80.6 to 119.8% (Table 3).
Carry over effect was found to be negligible for all substances. Regarding short term stability for 48 h at the autosampler and for 72 h at − 20 °C, it was found satisfactory, as for all the analytes it did not exceed 20%.
Detectability of drugs
The validated method was successfully applied to detection and quantification of drugs of abuse and prescribed drugs to earwax samples, confirming the presence of drugs of abuse into this biological specimen. We measured the concentrations of the selected analytes from 25 deceased drug users (24 men, 1 woman) 25–67 years of age.
In 20 of these cases, opiates were detected from blood and/or urine. Morphine was detected from cerumen in all of these cases (84%). In 3 cases, morphine was detected only from urine and not from blood, indicating a subacute drug use. In these 3 cases, morphine was also detected and quantified from cerumen. In one case, morphine was only detected from cerumen, indicated that the time range for the detectability of morphine from cerumen exceeds that from blood and also that from urine. Concentration of morphine varied from 1.00 to 487 ng/mg. Codeine (60%) was also detected to 15 cases from 1.56 to 37.7 ng/mg and 6-acetyl-morphine (60%) was also quantified in 15 cases in the range of 1.20–141 ng/mg. Similar to morphine, codeine and 6-acetyl-morphine were confirmed from cerumen in cases that these analytes were absent from blood and urine, confirming the wider detection time for the detectability of these analytes from cerumen. There was no correlation between the concentration found in blood and urine with the concentration in cerumen and subsequently no safe conclusion could be extracted regarding the variability in the concentrations in cerumen. Interindividual variability and also individual hygiene could partially explain the differences in concentrations found. In cases that opiates were detected from cerumen samples, noscapine was also detected and papaverine was detected in 18 of these cases suggested heroin intake. Also, in 15 cases, morhine-3-O-glucoronide was detected. The detection of morhine-3-O-glucoronide strongly indicates that the determination of opiates from cerumen cannot be a result of external contamination.
In cases 7, 9 and 24 where methadone and its metabolite EDDP were detected from blood and urine, methadone was also identified in cerumen samples and EDDP (8%) only in cases 7 and 24. Methadone (20%) was also detected from cerumen samples of cases 11 and 15, but was not detected from blood and urine.
Cocaine (n = 14, 56%) and its major metabolite benzoyl ecgonine (n = 14, 56%) were detected from cerumen samples in the concentration range of 1.14–6.86 and 1.00–64.8 ng/mg, but ecgonine methyl ester was not detected at all, following the same detection pattern as other biological specimens like hair and nails, where only trace amounts of ecgonine methyl ester were detected [23,24,25].
Benzodiazepines are usually co-abused with heroin and cocaine and as it shown in Supplementary Table 2, they can be quantified into earwax samples. Diazepam (n = 19, 76%) concentrations ranged from 1.23 to 12.9 ng/mg. Also, its major metabolite nordiazepam (n = 18, 72%) was detected in the range of 1.29–15.4 ng/mg. Oxazepam was only detected in eight cases (32%) and only in trace amounts or near the LOQ level (0.76–1.48 ng/mg). Temazepam (n = 13, 52%) concentrations varied between 1.00 ng/mg and 3.39 ng/mg. 7-Amino-flunitrazepam (52%) was detected in 13 cases (1.01–2.48 ng/mg), at most of them concentration lied below the LOQ of the validated method. Lorazepam (4%) was identified in one case at trace amounts. Alprazolam (n = 6, 24%) and bromazepam (n = 6, 24%) were detectable in the range of 1.05–2.06 ng/mg and 1.00–7.03 ng/mg, respectively.
Antidepressants citalopram (n = 4, 16%), venlafaxine (n = 1, 4%) and mirtazapine (n = 1, 4%) were detected from cerumen samples only in cases that these compounds were also present in blood and/or urine. Biperiden (n = 1, 4%), haloperidol (n = 3, 12%), quetiapine (n = 8, 32%) and olanzapine (n = 2, 8%) were also detected from cerumen samples. Sertraline and amitriptyline were not detected from cerumen samples in any of the 25 cases, but these analytes were also not detected from blood and urine. It should be mentioned that there was no significant difference at the concentration levels of all analytes between the right and the left side.
Discussion
According to our knowledge, this is the first described fully validated method for the detection of psychoactive drugs into cerumen samples. There are no published data for the determination of these analytes in cerumen samples. Meier et al. [12] analyzed 38 postmortem cerumen samples and detected drugs of abuse, stimulants (cocaine, amphetamines) and depressants of the central nervous system (opiates, benzodiazepines, Δ9- tetrahydrocannabinol), but they did not quantify these analytes. They performed LLE for the pretreatment of the samples, with ethyl acetate used as extraction solvent and LC–MS/MS applied for the screening of these drugs in cerumen. Shokry et al. [11] developed a method for the determination of antiepileptic and anxiolytic/antipsychotic drugs in cerumen from patients treated with a single antiepileptic or different multiple antiepileptic and anxiolytic/antipsychotic drug combinations. Methanol was used for the extraction of analytes of interest from cerumen and at this case an LC–MS/MS method was applied. In 2020, Meier et al. developed a method for the determination of 4-fluoroamphetamine in cerumen after controlled oral application [14]. At this work, they extracted the analyte with ethyl acetate followed by solid phase extraction (SPE) prior to LC–MS/MS analysis.
In this work, we established a simple, sensitive, straightforward LLE method, using methanol for the extraction of analytes, with no dryness step, and only 600 μL of solvent. Our results indicate that the developed method provides great extraction efficiencies, without interferences from the matrix, with low LOQs and satisfactory precision data. Finally, the method was successfully applied to 25 cerumen samples from deceased drug users.
Our results suggest a wider time frame for the detectability of some drugs of abuse from earwax samples, such as opiates, methadone, cocaine, benzoyl ecgonine and quetiapine since these analytes at several cases were only detected from cerumen and not from blood or urine. For these analytes, cerumen analysis could indicate past drug use but not cause of death, since there was no correlation between the blood and cerumen concentrations. This comes in accordance with the work of Meier et al. [12] who concluded that detection time window of cerumen exceeded that of urine but not this of hair. For benzodiazepines further studies should be performed since they were mainly detected in cases that they are present in blood and/or urine, perhaps due to their prolonged half-life. Citalopram, venlafaxine, haloperidol, olanzapine, biperiden and mirtazapine are only identified in cerumen if present in blood and/or urine, hence no safe conclusions, concerning the detection time frame of these substances, can be extracted without further studies. The simultaneous detectability of a drug in cerumen and blood can be explained by the fact that cerumen partly consists of sweat [6].
The detection of metabolites from cerumen and not only parent drugs suggest that the deposition at this biological specimen should result from excretory mechanisms which include circulating metabolites. Also, the detection of metabolites excludes the possibility of external contamination since in that case only parent drugs should be detected and not their metabolites.
Its ease of use, the minimum sample pretreatment and the fact that it is a biological sample that it is less prone to external contamination, due to its anatomical structure make it a useful specimen for the detection of drugs. Due to its no invasive collection it can also be preferred from sweat and saliva collection mainly in clinical toxicology for psychiatric patients who are unwilling to collaborate with the medical staff. The selection of cerumen as an alternative biological matrix overcomes the limitations of hair and nail analysis due to cosmetic manipulation and to external contamination and use of acetone and nail polish, respectively.
In cases of deceased at early stage of decomposition, cerumen can also be used in addition with hair and nail analysis. Cerumen seems to be a promising alternative biological matrix, that can be used both in clinical and forensic toxicology for the determination of a large number of drugs for a more comprehensive interpretation of the toxicological results.
Conclusion
We developed and fully validated a simple, quick and sensitive LLE method using LC-MS/MS analysis for the detection of 25 psychoactive drugs in cerumen, with low LOQs, and adequate precision, recovery and matrix effect data. It is the first time that these analytes are quantified in cerumen samples. Its application to 25 real samples shows that detection in cerumen could indicate past drug abuse. Additional studies with simultaneous analysis of hair and cerumen samples should be done to evaluate the exact time frame for drug detection in earwax.
References
Pragst F, Balikova MA (2006) State of the art in hair analysis for detection of drug and alcohol abuse. Clin Chim Acta 370(1–2):17–49
Solimini R, Minutillo A, Kyriakou C, Pichini S, Pacifici R, Busardo FP (2017) Nails in forensic toxicology: an update. Curr Pharm 23(36):5468–5479. https://doi.org/10.2174/1381612823666170704123126
Hartwig S, Auwärter V, Pragst F (2003) Effect of hair care and hair cosmetics on the concentrations of fatty acid ethyl esters in hair as markers of chronically elevated alcohol consumption. Forensic Sci Int 131(2–3):90–97. https://doi.org/10.1016/s0379-0738(02)00412-7
Schubert W (2013) Urteilsbildung in Der Fahreignungsbegutachtung: Beurteilungskriterien, 3rd edn. Kirschbaum, Bonn
Cappelle D, Yegles M, Neels H, Nuijs ALN, Doncke M, Maudens K, Covaci A, Crunelle CL (2014) Nail analysis for the detection of drugs of abuse and pharmaceuticals: a review. Forensic Toxicol 33(1):12–36. https://doi.org/10.1007/s11419-014-0258-1
Roeser RJ, Ballachanda BB (1997) Physiology, pathophysiology, and anthropology/epidemiology of human earcanal secretions. J Am Acad Audiol 8(6):391–400
Makino K, Amatsu M (1986) Epithelial migration on the tympanic membrane and external canal. Arch Otorhinolaryngol 243(1):39–42. https://doi.org/10.1007/BF00457906
Naiberg JB, RobinsonA KP, Hawke M (1992) Swirls, wrinkles and the whole ball of wax (the source of keratin in cerumen). J Otolaryngol 2:142–148
Guest JF, Greener MJ, Robinson AC, Smith AF (2004) Impacted cerumen: composition, production, epidemiology and management. QJM 97:477–488
Stoeckelhuber M, Matthias C, Andratschke M, Stoeckelhuber BM, Koehler C, Herzmann S, Sulz A, Welsch U (2006) Human ceruminous gland: ultrastructure and histochemical analysis of antimicrobial and cytoskeletal components. Anat Rec Part A 288:877–884. https://doi.org/10.1002/ar.a.20356
Shokry E, MarquesJG RPC, Pereira NZ, Filho NRA (2017) Earwax as an alternative specimen for forensic analysis. Forensic Toxicol 35(2):348–358. https://doi.org/10.1007/s11419-017-0363-z
Meier SI, Koelzer SC, Zsilavecz MS, Toennes SW (2017) Analysis of drugs of abuse in cerumen - correlation of postmortem analysis results with those for blood, urine and hair. Drug Test Αnal 9(10):1572–1585. https://doi.org/10.1002/dta.2177
Stepanov I, Feuer R, Jensen J, Hatsukami D, Hecht SS (2006) Mass spectrometric quantitation of nicotine, cotinine, and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol in human toenails. Cancer Epidemiol Biomarkers Prev 15:2378–2383
MeierSI SSP, Zsilavecz MS, Fernandes Perna ES, Theunissen EL, Ramaekers JG, Toennes SW (2020) Analysis of 4-fluoroamphetamine in cerumen after controlled oral application. Drug Test Anal 12:968–974. https://doi.org/10.1002/dta.2796
Shokry E, de Oliveira AE, Avelino MAG, de Deus MM, Pereira NZ, Filho NRA (2017) Earwax: an innovative tool for assessment of tobacco use or exposure. A pilot study in young adults. Forensic Toxicol 35(2):1–10. https://doi.org/10.1007/s11419-017-0370-0
Nicolaou AG, Stavrou IJ, Louppis AP, Constantinou MS, Kapnissi-Christodoulou C (2021) Application of an ultra-performance liquid chromatography-tandem mass spectrometric method for the detection and quantification of cannabis in cerumen samples. J Chromatogr A 1642:462035. https://doi.org/10.1016/j.chroma.2021.462035
Orfanidis A, Raikos N, Brousa E, Zangelidou E, Mastrogianni O (2021) Determination of lidocaine in postmortem whole blood samples after unsuccessful cardiopulmonary resuscitation. Seperations 8(8):117. https://doi.org/10.3390/separations8080117
Orfanidis A, Gika HG, Theodoridis G, Mastrogianni O, Raikos N (2021) A UHPLC-MS-MS method for the determination of 84 drugs of abuse and pharmaceuticals in blood. J Anal Toxicol 45(1):28–43. https://doi.org/10.1093/jat/bkaa032
Cao Z, Kaleta E, Wang P (2015) Simultaneous quantitation of 78 drugs and metabolites in urine with a dilute-and-shoot LC–MS-MS assay. J Anal Toxicol 39:335–346. https://doi.org/10.1093/jat/bkv024
Matuszewski BK, Constanzer ML, Chavez-Eng CM (2003) Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal Chem 75:3019–3030
Peters FT, Drummer OH, Musshoff F (2007) Validation of new methods. Forensic Sci Int 165:216–224
Scientific Working Group for Forensic Toxicology (SWGTOX) (2013) Standard practices for method validation in forensic toxicology. J Anal Toxicol 37(7):452–474
Garside D, Ropero-Miller JD, Goldberger BA, Hamilton WF, Maples WR (1998) Identification of cocaine analytes in fingernail and toenail specimens. J Forensic Sci 43(5):974–979
Engelhart DA, Jenkins AJ (2002) Detection of cocaine analytes and opiates in nails from postmortem cases. J Anal Toxicol 26:489. https://doi.org/10.1093/jat/26.7.489
Phipps RJ, Smith JJ, Darwin WD, Cone EJ (2008) Handbook of analytical separations. Elsevier B.V., Amsterdam, p 6. https://doi.org/10.1016/S1567-7192(06)06002-5
Acknowledgements
The authors wish to acknowledge the technical employees of the Forensic Service of Thessaloniki for providing the biological samples.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in this study were in accordance with the ethical standards of the national committee and with the 1964 Declaration of Helsinki. Biological fluids (blood, urine) and cerumen collection from decedents was made by judicial authorities, and the samples were sent to our institute for toxicological analysis at their request. This article does not contain any studies with living human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Mastrogianni, O., Orfanidis, A., Brousa, E. et al. Development and validation of an ultra-performance liquid chromatography–tandem mass spectrometric method for the determination of 25 psychoactive drugs in cerumen and its application to real postmortem samples. Forensic Toxicol 41, 94–104 (2023). https://doi.org/10.1007/s11419-022-00640-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11419-022-00640-y