
Abstract
Purpose
Many biotic and abiotic factors influence the structural and functional diversity of microbial communities in the rhizosphere. This study aimed to understand the dynamics of fungal community in the soybean rhizosphere during soybean growth and directly compare the influence of abiotic and biotic factors in shaping the fungal communities across different growth periods.
Materials and methods
High-throughput sequencing based on internal transcribed spacer (ITS) region, quantitative PCR, and statistical analysis approaches were used to measure the fungal community structure, abundance, and dynamic changes of 63 rhizosphere soil samples which were taken from different fertilization regimes and rhizobium inoculation treatments during three soybean growth stages.
Results and discussion
Among the taxa examined, more than 16 fungal classes were detected from the 21 soil samples. Sordariomycetes was the most abundant class, followed by Dothideomycetes, Agaricomycetes, and Eurotiomycetes. Soybean growth stage was the most important factor determining the diversity patterns of the fungal communities. Fungal community diversity was closely related to the base-fertilizer application, and fungal community richness was associated with rhizobium inoculation. Beta diversity of the fungal community based on the Bray-Curtis distance was significantly related to plant growth stage. Network analysis showed that mutual cooperation among fungal taxa became more intimate during the plant growth.
Conclusions
Compared with edaphic properties, plant growth stage was the dominant factor in determining soil fungal community dynamics. Base-fertilizer and rhizobium inoculation affected the alpha diversity of the soil fungi.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Fungi play important and extremely complex roles in the soil biogeochemical cycle and during plant growth and are fundamental to the soil ecosystem. The beneficial effects of soil fungi include organic matter decomposition, breakdown of metabolic by-products and agrochemicals, and enhancement of soil element bioavailability (Broeckling et al. 2008). For example, mycorrhiza-mediated competition between plants and decomposers can drive soil carbon storage (Averill et al. 2014) and mycoparasitic fungi are able to improve plant resistance to fungal pathogens (Lorito et al. 1998). An improved knowledge of the diversity and structure of fungal communities in rhizosphere soils can lead to a better understanding of their roles in soil ecosystems.
A number of studies have shown that fungal community composition and abundance shift in response to soil pH, total carbon (TC) content, land-use patterns, and other environmental factors (e.g., soil types, vegetation types, and soil animals) (Christensen 1989; Vandenkoornhuyse et al. 2002; Anderson and Cairney 2004; Berg and Smalla 2009; Liu et al. 2015). Ectomycorrhizal fungal richness is most strongly related to host plant species diversity and high soil pH; saprotroph richness is positively related to mean annual precipitation; and pathogen richness is negatively related to latitude, but positively related to nitrogen availability (Wardle and Lindahl 2014). Many biotic and abiotic factors are thought to influence the structural and functional diversity of microbial communities in the rhizosphere (Drenovsky et al. 2004; Fierer and Jackson 2006; Singh et al. 2009). However, there have been few studies on fungal communities in the rhizosphere considering the influence of growth periods.
This study aimed to (1) investigate the presence and structural diversity of fungi in the soybean rhizosphere during plant growth; (2) examine the variability of fungal community structure during soybean growth and the nodulation process; and (3) directly compare the influence of abiotic and biotic factors in fungal communities across different growth stages. To do this, we collected soils after the application of different fertilization and rhizobium inoculation treatments and used Miseq pyrosequencing of the internal transcribed spacer (ITS) region and quantitative PCR (qPCR) to determine the structure and relative abundance of the soil fungal communities.
2 Materials and methods
2.1 Soil sampling
Soil samples were collected from the Shofine Seed Industry Academic Workstation (116° 37′ 61.19″ E, 35° 44′ 16.69″ N), located in Jiaxiang County, Shandong Province, middle East China. The plots at Shofine were selected for sampling because it was an important rhizobium research center at the China Agricultural University, Beijing, China, and had been running rhizobium selection tests for 2 years. The soybean (var. Shannong 20) was subjected to one of seven experimental treatments with three replicates per treatment (Table 1). The base-fertilizer (BF) treatment was basic fertilized 225 kg/ha with NPK compound fertilizer 15-15-15 before sowing the seeds. Sinorhizobium fredii S6 was inoculated by seed dressing and liquid spraying combined technology as a rhizobium inoculation (RI) treatment. At the early flowering stage, 150 kg/ha urea was further applied in the top dressing (TD) treatment soil. Samples were collected from the seedling stage (22 June, 2014), flowering stage (4 Aug. 2014), and mature stage (29 Sept. 2014). This yielded a total of 63 samples (7 treatments, 3 stages, 3 replicates). Each soil sample was passed through a 2.0-mm sieve, partial fresh sample stored at 4 °C for physical and chemical analyses and others stored at −80 °C after freeze-drying for DNA extracting.
2.2 Soil biogeochemical properties
All samples were subjected to physical and chemical analyses to determine their major soil characteristics (Table 1). Soil pH and electrical conductivity (EC) were determined at a ratio of 1:5 (soil to water, w/w). Soil organic carbon (SOC) was determined using the K2Cr2O7 oxidation method (Sparks et al. 1996), while total nitrogen (TN) was determined using a Vario EL III analyzer (Elementar Analysensysteme GmbH, Hanau, Germany). Soil ammonium (NH4 +-N) and nitrate (NO3 −-N) were extracted with 2 M KCl and determined by a continuous flow analyzer (SAN++; Skalar, Breda, Holland). Soil available phosphorus was determined using the Olsen method (Olsen 1954), and soil available potassium was extracted with 0.5 M ammonium acetate and determined by an atomic absorption spectrophotometer (ZEEnit700P, Analytik Jena AG, Germany). All these analyses were carried out in triplicate.
2.3 DNA extraction and qPCR analysis of the ITS region
Total DNA was extracted from 0.25 g soil samples using a MoBio Powersoil® DNA Isolation Kit (MoBio Laboratories, Carlsbad, CA, USA) according to the manufacturer’s protocol. The fungal abundances were quantified by qPCR that targeted the fungal ITS1 region. The ITS1 region was analyzed using ITS5 (GGAAGTAAAAGTCGTAACAAGG) and ITS2 (GCTGCGTTCTTCATCGATGC) primers, according to White et al. (White et al. 1990). The reactions were performed on an iQTM5 Thermocycler (Bio-Rad, USA) in a volume of 25 μL that contained 1 μL of extracted DNA, 12.5 μL of Power SYBR green master mix (Takara Biotechnology, Dalian, China), and 0.25 μM of each primer. The following thermal program was used for amplification: 95 °C for 5 min, 40 cycles of 95 °C for 45 s, 50 °C for 30 s, and 72 °C for 45 s, followed by a plate read at 83 °C. The specificity of the amplification products was confirmed by melting curve analysis, and the expected sizes of the amplified fragments were checked in a 1.5 % agarose gel stained with GoldView (GV). To prepare standard curves, the ITS sequences were amplified and ligated to a pGEM-T Easy vector (Promega, USA) and transformed into Escherichia coli JM109 cells. Positive clones containing the target gene insert were sequenced and the most abundant one was used for plasmid DNA extraction. After measuring the DNA concentration with a NanoPhotometer® P-Class (Implen GmbH, Germany), the purified plasmid DNA was diluted serially in 10-fold steps and subjected to real-time PCR in triplicate to generate an external standard curve. Threshold values obtained from sample amplification were interpolated from the standard curve to determine the number of fungi found per gram of soil.
2.4 Barcoded pyrosequencing and processing of the pyrosequencing data
The fungal community was analyzed using the ITS1 region, which is commonly used to pyrosequence environmental samples (Bates et al. 2013; Meiser et al. 2014; Liu et al. 2015). A 50 μL PCR mixture containing 25 μL premix (Takara Biotechnology, Dalian, China), 1 μL of each primer (10 μM), 2 μL DNA template (about 10 ng), and 21 μL ddH2O was used. The thermal cycling parameters for qPCR were the same as those mentioned above. The PCR products were purified using a Wizard SV Gel and PCR Clean-up system (Promega, Madison, WI, USA) and then sequenced on the MiSeq platform (Illumina, San Diego, CA, USA) at Novogene, Beijing, China.
A 97 % similarity was used in the operational taxonomic unit (OTU) definition for estimating the alpha and beta diversity. The Shannon index, Simpson index, chao1, and phylogenetic diversity (PD) whole tree values were calculated to compare the alpha diversities. The Bray-Curtis distances were introduced as the beta diversity metrics. An Unweighted Pair Group Method with Arithmetic Mean clustering (UPGMA) based on the Bray-Curtis distances was performed in Quantitative Insights Into Microbial Ecology (QIIME) software to compare the pair-wise community similarity (Smit et al. 1999).
2.5 Statistical analysis
Statistical significance was assessed using SPSS 19.0 software (SPSS Inc., Chicago, IL, USA). One-way analysis of variance (ANOVA) was used to assess the differences among soil factors. Redundancy analysis (RDA) (Canoco 4.5 for windows) was used to explore the relationships between the samples and environmental factors (Buee et al. 2009). The permutational multivariate analysis of variance analyzed whether the OTUs in different treatments were statistically different by using the adonis function in the vegan package of the R platform with 999 permutations. A network analysis was performed with a CoNet Cytoscape plug-in (Becklin et al. 2012), based on 97 % OTU identity, in order to explore the linkage among different fungal taxa.
3 Results
3.1 Variations in environmental factors among treatments
A Spearman’s correlation matrix was used to examine the correlations among the fungal abundances, BF, RI, TD, and edaphic properties, including pH, EC, SOC, TN, available phosphorus, available potassium, NH4 +, and NO3 − (Table 1). No significant relationships were observed between RI and any soil properties. TD had significantly positive correlations with SOC (r 2 = 0.53, P < 0.05), TN (r 2 = 0.51, P < 0.05), and available phosphorus (r 2 = 0.50, P < 0.05). Fungal gene abundances were significantly negatively related to soybean growth stage (r 2 = 0.54, P < 0.05).
3.2 Fungal community composition, abundance and diversity
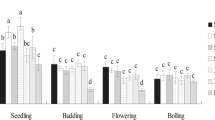
There were 6,034,418 high-quality ITS region sequences obtained through the high-throughput sequencing of 63 soil samples during the fungal community analysis, and 95,784 sequences on average were assigned to each sample. The fungal community compositions were profiled according to their relative abundance at the taxonomic class level (Fig. 1). Among the taxa examined, more than 16 fungal classes were detected from the 21 experimental soils. Sordariomycetes was the most abundant class with relative abundances ranging from 57.9 to 81.87 %, followed by Dothideomycetes, Agaricomycetes, and Eurotiomycetes with relative abundances ranging from 2.13 to 10.44 %, from 0.39 to 14.64 %, and from 0.22 to 2.7 %, respectively. In addition, numerous sequences were grouped as unclassified fungi and others with relative abundances that varied from 3.78 to 7.28 % and from 17.65 to 25.5 %, respectively. Quantitative PCR assays revealed that the fungal ITS region abundance of all soil samples ranged from 4.15 × 106 to 3.22 × 107 copies per gram soil (Table 1).

Relative abundance of dominant classes of fungi in the different treatments
3.3 The influence of treatments and environmental effects on fungal communities
RDA was used to further investigate the influences of different soil physical and chemical properties on community assemblages (Fig. 2). The first axis explained 80.85 % and the second axis accounted for 1.43 % of the variance in the fungal communities. Among all the soil geochemical factors examined, soil EC (32.14 %), NH4 +-N (4.15 %), NO3 −-N (1.64 %), and available phosphorus (3.11 %) were found to have significant effects (P < 0.05) on the fungal diversity of the soil samples. However, a relationship between soil SOC and fungal communities was not found. For the fungal communities, EC, available phosphorus, and NO3 −-N were positively correlated with soybean seedling stage, and NH4 +-N was positively correlated with the soybean flowering and mature stages.

Relationships (redundancy analysis) between different soil physicochemical factors and fungal communities in the three plant growing stages. The triangles represent samples taken at the seedling stage, the stars represent samples taken at the flowering stage, and the circles represent samples taken at the mature stage
We also used two-way permutational multivariate analysis of variance to explore the effects of different treatments and soybean growth stages on fungal beta diversity patterns. The fungal community composition in soil samples with rhizobia inoculation was significantly different from the non-inoculated soils (P < 0.05). The effects of base-fertilization on the soil fungal communities and growth stage were also very significant compared to soil samples that had received no base-fertilization (P < 0.001) (Table S1, Electronic Supplementary Material).
We used an UPGMA hierarchical clustering procedure with Bray-Curtis distances to account for the phylogenetic beta diversity of fungal communities. The three main clusters were relatively consistent with soybean growth stage and experimental treatment, and these were marked with different colors (Fig. 3). The UPGMA tree showed that fungal communities from the first batch of soil samples (seedling stage) were more likely to be clustered together, while the second batch (flowering stage) and third batch (mature stage) samples with base-fertilizer taken later in the plant growing season showed clustering that crossed over. The third main cluster was consistent with mature stage samples (M5, M6, and M7) that had received no base-fertilizer.

UPGMA dendrogram constructed from a Bray-Curtis distances matrix of the fungal ITS region. The three main clusters colored blue, red, and purple represent each plant growth stage of seedling, flowering, and mature stage, respectively
3.4 Network analysis of soil fungal communities
The OTU networks were used to explore the relationships among different fungal taxa. In this study, three networks from three growth stages were analyzed (Fig. 4). The network at the seedling stage revealed 38 significant correlations (edges) and 29 OTUs (nodes) (Fig. 4a), and the network at the flowering stage had 77 significant correlations (edges) and 70 OTUs (nodes) (Fig. 4b), while the network at the mature stage had 109 significant correlations (edges) and 153 OTUs (nodes) (Fig. 4c). Most of the nodes interacted cooperatively (positive correlations). Furthermore, mutual cooperation among fungal taxa became more intimate as the plant growing.

Network analysis on the fungal communities in the soils for the three plant growth stages (a, Seedling stage. b, Flowering stage. c, Mature stage). Node size represents the eigenvector centrality score. The larger the nodes, the more important they are in the network. The green line represents a positive correlation, whereas the red line represents a negative correlation
4 Discussion
Fungi are critical in soils and perform a range of important ecological functions, such as pathogenesis, symbiosis, and decomposition. They also drive nutrient and carbon cycling processes (Christensen 1989; Anderson and Cairney 2004). In this study, qPCR and Miseq high-throughput sequencing were used to analyze the abundance and diversity of fungal communities. The data showed that Ascomycota was the most abundant phylum, followed by Basidiomycota. Similar results were found for fungal communities growing in wheat rhizosphere soils, which mainly included Basidiomycota, Zygomycota, and Ascomycota (Smit et al. 1999). Likewise, Ascomycota and Basidiomycota were the most abundant operational taxonomic units in forest soils (Buee et al. 2009).
Alpha diversity, including richness, evenness, and phylodiversity, also interacted with different tillage management processes (Table S2, Electronic Supplementary Material). The evenness results (Shannon and Simpson indexes) showed significantly positive correlations with base-fertilizer (P < 0.05), while richness (Chao1, P < 0.01) and phylodiversity (PD_whole_tree, P < 0.05) decreased significantly after rhizobium inoculation. Top dressing had no significant influence on alpha diversity. We also found significant differences between the seedling, flowering, and maturity stages when abundance and β-diversity were used as indicators. Studies of rhizosphere fungi have previously recognized plant varieties, root exudates, and soil type, as major drivers of fungal community compositions (Broeckling et al. 2008; Berg and Smalla 2009; Becklin et al. 2012). However, few studies have focused on plant growth period. Here, we identified soybean growth stage as the most important factor determining the diversity patterns in fungal communities.
Considerable amounts of detailed information about the characteristics of the fungal communities in rhizosphere soils at three different soybean growth stages were obtained through network analysis, which is a systems biological approach that explores the relationships between different taxa by Miseq high-throughput sequencing (Raes and Bork 2008) and their distinct modular structures (Newman 2006). The high levels of Agaricales at the seedling stage indicated that they were frequently related to other taxa, and Mortierellales and Hypocreales showed more significant connections with other species at the flowering stage, while the network at the mature stage had the most modules and interactions. A possible reason for these changes was that the rhizobium inocula adapted to the soil environment and built a good co-existence relationship with indigenous microorganisms.
In addition, plants have a strong influence on the diversity and composition of microbial communities over the vegetative period. The composition of fungal communities in forest soils is largely determined by the dominant trees (Jover et al. 2014). A previous study also reported that rhizosphere fungal communities were dependent on the varieties of soybean grown in a field (Prigent et al. 2005). However, Sugiyama et al. (2014) suggested that large proportions of the rhizosphere fungal communities remained stable during soybean growth when the changes in the OTU relative abundances were compared. Similarly, the fungi in maize rhizosphere soil were also found to be little affected by maize root exudates during the corn growing season. In this study, an important finding was that the distribution patterns of the fungal communities at three stages were clearly different. The effects of plants on fungi are greater than the influences of fertilization and inoculation.
Although biotic factors explained the majority of variation in fungal community composition, abiotic factors cannot be entirely discounted. Nodulated soybean can enhance the rhizosphere priming effects on soil organic matter decomposition (Fu and Cheng 2002; Cheng et al. 2003) because the ammonia released from the soybean rhizosphere serves as both an energy substrate and an N source and thus promotes microbial turnover and SOM decomposition (Zhu and Cheng 2012). Furthermore, decomposition of SOC is the main process governing the release of CO2 into the atmosphere from terrestrial systems. Fungi are the major decomposers of organic matter (Holland and Coleman 1987) and the abundances and diversities of fungi at the flowering and mature stages increased significantly, whereas the stages from flowering to maturity were the most active period for soybean nodulation and nitrogen fixation.
5 Conclusions
In conclusion, this study analyzed fungal diversity and how the biotic and abiotic factors affected their diversity. Compared with edaphic properties controlling the soil fungal community patterns, plant growth stage was the dominant factor in determining soil fungal community dynamics. Fungal community diversity was closely related to base-fertilizer application, and fungal community richness was associated with rhizobium inoculation. Our results raise questions about the possible roles and ecological implications of fungal diversity associated with plant and environmental factors. Further studies need to quantify the fungal effects on soil biogeochemical cycling of nutrients and soybean production.
References
Anderson IC, Cairney JW (2004) Diversity and ecology of soil fungal communities: increased understanding through the application of molecular techniques. Environ Microbiol 6:769–779
Averill C, Turner BL, Finzi AC (2014) Mycorrhiza-mediated competition between plants and decomposers drives soil carbon storage. Nature 505:543–545
Bates ST, Ahrendt S, Bik HM, Bruns TD, Caporaso JG, Cole J, Dwan M, Fierer N, Gu D, Houston S (2013) Meeting report: fungal ITS workshop (October 2012). Stand Genomic Sci 8:118
Becklin KM, Hertweck KL, Jumpponen A (2012) Host identity impacts rhizosphere fungal communities associated with three alpine plant species. Microbial Ecol 63:682–693
Berg G, Smalla K (2009) Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere. FEMS Microbiol Ecol 68:1–13
Broeckling CD, Broz AK, Bergelson J, Manter DK, Vivanco JM (2008) Root exudates regulate soil fungal community composition and diversity. Appl Environ Microbiol 74:738–744
Buee M, Reich M, Murat C, Morin E, Nilsson RH, Uroz S, Martin F (2009) 454 Pyrosequencing analyses of forest soils reveal an unexpectedly high fungal diversity. New Phytol 184:449–456
Cheng W, Johnson DW, Fu S (2003) Rhizosphere effects on decomposition. Soil Sci Soc Am J 67:1418–1427
Christensen M (1989) A view of fungal ecology. Mycologia 81:1–19
Drenovsky R, Vo D, Graham K, Scow K (2004) Soil water content and organic carbon availability are major determinants of soil microbial community composition. Microbial. Ecol 48:424–430
Fierer N, Jackson RB (2006) The diversity and biogeography of soil bacterial communities. Proc Natl Acad Sci 103:626–631
Fu S, Cheng W (2002) Rhizosphere priming effects on the decomposition of soil organic matter in C4 and C3 grassland soils. Plant Soil 238:289–294
Holland E, Coleman DC (1987) Litter placement effects on microbial and organic matter dynamics in an agroecosystem. Ecology 2:425–433
Jover LF, Effler TC, Buchan A, Wilhelm SW, Weitz JS (2014) The elemental composition of virus particles: implications for marine biogeochemical cycles. Nature. Rev Microbiol 12:519–528
Liu J, Sui Y, Yu Z, Shi Y, Chu H, Jin J, Liu X, Wang G (2015) Soil carbon content drives the biogeographical distribution of fungal communities in the black soil zone of Northeast China. Soil Biol Biochem 83:29–39
Lorito M, Woo SL, Fernandez IG, Colucci G, Harman GE, Pintor-Toro JA, Filippone E, Muccifora S, Lawrence CB, Zoina A (1998) Genes from mycoparasitic fungi as a source for improving plant resistance to fungal pathogens. Proc Natl Acad Sci 95:7860–7865
Meiser A, Bálint M, Schmitt I (2014) Meta-analysis of deep-sequenced fungal communities indicates limited taxon sharing between studies and the presence of biogeographic patterns. New Phytol 201:623–635
Newman ME (2006) Modularity and community structure in networks. Proc Natl Acad Sci 103:8577–8582
Olsen SR (1954) Estimation of available phosphorus in soils by extraction with sodium bicarbonate. Circular 939. United States Department Of Agriculture, Washington
Prigent M, Leroy M, Confalonieri F, Dutertre M, DuBow MS (2005) A diversity of bacteriophage forms and genomes can be isolated from the surface sands of the Sahara Desert. Extremophiles 9:289–296
Raes J, Bork P (2008) Molecular eco-systems biology: towards an understanding of community function. Nat Rev Microbiol 6:693–699
Singh BK, Dawson LA, Macdonald CA, Buckland SM (2009) Impact of biotic and abiotic interaction on soil microbial communities and functions: a field study. Appl Soil Ecol 41:239–248
Smit E, Leeflang P, Glandorf B, van Elsas JD, Wernars K (1999) Analysis of fungal diversity in the wheat rhizosphere by sequencing of cloned PCR-amplified genes encoding 18S rRNA and temperature gradient gel electrophoresis. Appl Environ Microbiol 65:2614–2621
Sparks DL, Page A, Helmke P, Loeppert R, Soltanpour P, Tabatabai M, Johnston C, Sumner M (1996) Methods of soil analysis. Part 3-chemical methods. Soil Science Society of America. American Society of Agronomy, Madison
Sugiyama A, Ueda Y, Takase H, Yazaki K (2014) Pyrosequencing assessment of rhizosphere fungal communities from a soybean field. Can J Microbiol 60:687–690
Vandenkoornhuyse P, Baldauf SL, Leyval C, Straczek J, Young JPW (2002) Extensive fungal diversity in plant roots. Science 295:2051–2051
Wardle DA, Lindahl BD (2014) Disentangling global soil fungal diversity. Science 346:1052–1053
White TJ, Bruns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR protocols: a guide to methods and applications 18:315–322
Zhu B, Cheng W (2012) Nodulated soybean enhances rhizosphere priming effects on soil organic matter decomposition more than non-nodulated soybean. Soil Biol Biochem 51:56–65
Acknowledgments
This work was supported by the National Science Foundation of China (Grant No. 41301265) and the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB15020200).
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Yanfen Wang
Electronic supplementary material
ESM 1
(DOCX 22 kb)
Rights and permissions
About this article

Cite this article
Han, LL., Wang, JT., Yang, SH. et al. Temporal dynamics of fungal communities in soybean rhizosphere. J Soils Sediments 17, 491–498 (2017). https://doi.org/10.1007/s11368-016-1534-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11368-016-1534-y