
Abstract
Terminal sialic acid residues are present on most glycoproteins and glycolipids, but levels of sialylation are known to change in the brain throughout the lifespan as well as during disease. Sialic acids are important for numerous cellular processes including cell adhesion, neurodevelopment, and immune regulation as well as pathogen invasion into host cells. Neuraminidase enzymes, also known as sialidases, are responsible for removal of terminal sialic acids in a process known as desialylation. Neuraminidase 1 (Neu1) cleaves the α-2,6 bond of terminal sialic acids. Aging individuals with dementia are often treated with the antiviral medication oseltamivir, which is associated with induction of adverse neuropsychiatric side effects; this drug inhibits both viral and mammalian Neu1. The present study tested whether a clinically relevant antiviral dosing regimen of oseltamivir would disrupt behavior in the 5XFAD mouse model of Alzheimer’s disease amyloid pathology or wild-type littermates. While oseltamivir treatment did not impact mouse behavior or modify amyloid plaque size or morphology, a novel spatial distribution of α-2,6 sialic acid residues was discovered in 5XFAD mice that was not present in wild-type littermates. Further analyses revealed that α-2,6 sialic acid residues were not localized the amyloid plaques but instead localized to plaque-associated microglia. Notably, treatment with oseltamivir did not alter α-2,6 sialic acid distribution on plaque-associated microglia in 5XFAD mice which may be due to downregulation of Neu1 transcript levels in 5XFAD mice. Overall, this study suggests that plaque-associated microglia are highly sialylated and are resistant to change with oseltamivir, thus interfering with microglia immune recognition of and response to amyloid pathology.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Aging is the biggest risk factor in the pathogenesis of Alzheimer’s disease (AD) and other related dementias. Older adults, including AD patients, are more vulnerable to viral infections such as influenza A [29]. Many people are treated for influenza with antiviral medications [19] such as oseltamivir (brand name Tamiflu). Oseltamivir inhibits viral neuraminidase 1 (Neu1), thus blocking viral invasion into host cells, and reduces viral load and symptom duration [19]. While oseltamivir has been commercially available for decades and is generally well-tolerated, there are reports of severe adverse neuropsychiatric side effects, both acute and delayed, in patients treated with oseltamivir including psychosis, confusion, aggression, suicidal risk, and standing instability [46]. In one case study, an aged patient with no family history of psychiatric disorders developed hallucinations and delirium, within 4 days of oral oseltamivir treatment [6]. Other studies have shown greater incidences of both acute and delayed adverse neuropsychiatric side effects in patients of advanced age or AD treated with oseltamivir for flu symptoms compared to non-AD patients [16]. Therefore, we hypothesized that these adverse neuropsychiatric side effects could indicate an interaction between AD pathology and the mechanism of oseltamivir.
Oseltamivir is a viral neuraminidase 1 (Neu1) inhibitor that can also inhibits the activity of host mammalian Neu1. Neuraminidase enzymes specifically hydrolyze terminal sialic acid (SA) post-translational modifications (PTMs) attached to glycosylated proteins and lipids in a process called desialylation [3, 39]. Reports suggest Neu1 acts preferentially on glycoproteins [45]. There are several classes of mammalian neuraminidase enzymes that act on specific targets of sialylated glycoconjugates. Specifically, Neu1 acts on sialylated glycopeptides and oligosaccharides by targeting the α-2,6 SA bond type and α-2,3 SA bonds, although to a lesser extent. Neu1 is expressed at the highest levels in the kidney, pancreas, liver, lungs, and ubiquitously in the brain with strong expression in the hippocampus and cortex [39].
SA PTMs belong to the N-acetylneuraminic acid family with a nine-carbon backbone attached to terminal ends of glycans [23]. The functions of SA PTMs are multi-fold with impacts in cell adhesion, neurodevelopment, and immune regulation. The biological significance of α-2,6 SA bond type is the relation to microglia activity and AD. This SA bond type is a target for Neu1 and serves as a ligand for inhibitory receptors that modulate microglia activation [25, 36]. Notably, SA PTMs undergo changes during aging that may mediate the development of AD. The cerebrospinal fluid (CSF) of patients diagnosed with mild cognitive impairment (MCI) is enriched for galactose SA metabolism compared to non-MCI patients [15, 47, 48], and greater declines in CSF SA are associated with the conversion of MCI to AD [35]. In AD patients, Aβ plaques contain sialylated glycoproteins and glycolipids, specifically sialylated clusterin protein and amyloid precursor protein (APP) [40, 42]. Further, Aβ binds with high affinity to SA clustered on cell surfaces [7]. The innate immune cell in the brain, microglia, play a multifaceted role in the pathogenesis of AD [44] and are partly regulated by SA. Microglia use cell surface sialylation as a checkpoint for innate immune response [21, 22]. Microglia express inhibitory sialic acid-binding immunoglobulin-like lectins (Siglecs) that detect SA and trigger specific downstream pathways [4]. Desialylation of microglia further enhances their immune response [2]. Taken altogether, these data suggest that modulation of brain Neu1 could potentially mediate adverse neuropsychiatric side effects of oseltamivir via SA on either Aβ plaques or on microglia cell surface.
To test whether a clinically-relevant antiviral dosing regimen of oseltamivir could mediate neuropsychiatric side effects via modulation of SA in the brain, we orally administered oseltamivir to a cohort of 12-month-old 5XFAD mice and wild-type littermates twice a day for five days and measured effects on behavior, neuropathology, and spatial organization of α-2,6 SA in the brain. Notably, previous studies on SA spatial distribution within the brain in AD have been limited, and it remains unclear which SA PTMs are localized to AD pathology [31]. We discovered a novel spatial distribution of α-2,6 SA residues in 5XFAD mice that was not present in wild-type littermates, although oseltamivir treatment did not impact mouse behavior or modify amyloid plaque size or morphology. Intriguingly, we found that α-2,6 SA is primarily localized to microglia in the Aβ plaque microenvironment, suggesting that α-2,6 sialylation of plaque-associated microglia may be involved in microglia-plaque interactions during AD.
Materials and methods
Animals
All animal experiments were approved by the UT Health San Antonio Institutional Animal Care and Use Committee in alignment with the Guide for the Care and Use of Laboratory Animals. We used hemizygous 5XFAD mice on congenic C57BL/6 J background and wild-type (WT) littermates, with founders obtained from Jackson Laboratories (MMRRC #034848-JAX). 5XFAD mice express human APP harboring three mutations associated with familial AD (Swedish [K670N/M671L], Florida [I716V], and London [V717I] mutations) and human presenilin-1 (PSEN1) with M146L and L286V mutations [33]. 5XFAD hemizygotes and WT littermate mice were generated by crossing hemizygous 5XFAD mice with C57BL/6 J mice (JAX stock #000,664). In this study, male and female mice approximately 12 months old were used. Mice were handled once per week for 1 min minimum starting at 8–12 weeks of age to reduce stress while in behavioral testing. All mice in this study were housed in ventilated cages with a 14-h light and 10-h dark cycle (7 am–9 pm) in a climate-controlled room. Food and water provided ad libitum, and mice were group housed with a maximum of 5 mice per cage. Mice were genotyped by gathering DNA from tail cuts and identified by ear notches. Genotyping was performed by polymerase chain reaction (PCR) to validate 5XFAD mutations (mutant prime sequence 5′3′ CGG GCC TCT TCG CTA TTA C; common prime sequence ACC CCC ATG TCA GAG TTC CT; and wild-type reverse prime sequence TAT ACA ACC TTG GGG GAT GG) and PCR products visualized by gel electrophoresis.
Drug administration
This study used a total of 56 mice (n = 18 5XFAD females; n = 13 5XFAD males; n = 12 WT females; n = 13 WT males). Mice were treated with oral (po) oseltamivir phosphate (Sigma Aldrich SML1606-100MG) dissolved in ultrapure water at 10 mg/kg or ultrapure water vehicle twice per day (bid) for 5 days with an 8-h window between doses. This dose was selected because reports show 10 mg/kg po dosing delivered adequate bioavailability of the drug and was in alignment with human dosing of 75 mg twice daily [51, 53]. This treatment dose alleviates viral symptoms in mice inoculated with influenza A, and this dose is partially metabolized in the brain [17, 53]. To control for different body weights based on sex, the average body weight per sex was used to calculate drug concentration. Male mice were an average weight of 0.029 kg using this; the drug concentration was 2.9 mg/ml with a drug dose of 10 mg/kg. Female mice weighed an average of 0.023 kg; the drug concentration was 2.3 mg/ml with a drug dose of 10 mg/kg. Oseltamivir was administered with a total volume of 100 μL using a 1-mL syringe and metal size 20 gavage tube.
Behavior
All mice were assessed through two behavioral paradigms (Fig. 2a). Acute behavioral changes were measured on day 3 of treatment. Chronic or prolonged behavioral changes were assessed starting on day 7 and continued to day 9. Mice were acclimated to the behavior room in their home cages at 7 am before behavior started at 8 am. All behavior was completed under standard room lighting, and Noldus Ethovision 14 was used to record and score behavior. All behavioral apparatuses were cleaned with 70% ethanol between animals. GraphPad Prism (version 9.3.1) software was used for all statistical analyses.
Open field (OF)
OF was used to probe for anxiety-like behaviors or gross motor deficits induced by oseltamivir. OF testing was performed on days 3 and 7 of oseltamivir treatment to detect any acute or delayed behavioral changes, respectively. On day 3, mice were PO dosed in the morning and then moved to the behavior room for 1 h to acclimate before OF testing began. Mice were placed in the center of the testing arena (42 L × 20 W x 20H) surrounded by an opaque barrier and explored for 10 min. After 10 min, mice were returned to their home cage. Velocity as a measure of motor function and time spent in the center of the arena as a measure of anxiety levels were recorded for analysis. Anxiety-like behaviors were assessed using a 2-way ANOVA (treatment × genotype) on time spent in the center of the arena. All behavioral analyses were conducted using standard error of the mean (SEM). We analyzed differences between testing days with a 3-way repeated measures ANOVA on OF velocity data (day × treatment × genotype). A second 3-way repeated measures ANOVA (minute × treatment × genotype) was used to look at habituation to the testing environment. All group comparisons were made with post-hoc Bonferroni’s correction.
Novel object recognition (NOR)
NOR was used to assess recognition memory changes induced by oseltamivir treatment. Upon completion of drug administration and OF testing (which served as an acclimation trial for NOR), mice were trained in a learning trial, during which two identical objects (3.8 × 5 cm plastic candles, familiar object) were placed at one end of the arena approximately 3.8 cm away from the wall and 13 cm away from the other object. Mice were placed at opposite ends of the arena, and exploration behavior was recorded for 10 min before being returned to their home cages. The next day, testing was repeated with one of the familiar objects replaced with a novel object (3 × 3 × 3 cm cube). Object exploration was defined as either direct or indirect (nose pointed toward object within a 3.8-cm diameter perimeter) nose interaction with an object. Total exploration time was defined as the summation of nose interaction and perimeter interaction. Recognition memory was quantified via discrimination index (DI) [(time spent with novel object – time spent with familiar object)/(total exploration time of familiar + novel object)]. Velocity and time spent in the center of the arena were also recorded. Mice that did not explore objects for more than 20 s in the learning and/or testing trial were excluded from analysis. To evaluate differences in locomotion between testing days, a 3-way repeated measures ANOVA on OF velocity data (day × treatment × genotype) was used. Changes in DI were assessed a 2-Way ANOVA (treatment × genotype).
Euthanasia and tissue processing
Mice were euthanized in accordance with American Veterinary Medical Association guidelines using isoflurane followed by cervical dislocation once mice were unresponsive to toe pinch. Brains were hemisected with the left hemibrain post-fixed in 10% neutral buffered formalin (Leica cat. #3,800,540) for 24–36 h and the right hemibrain frozen. Fixed tissue was sectioned coronally, processed for 8 h (Leica ASP300S Tissue Processor), paraffin-embedded into tissue blocks using Histoplast Paraffin wax (ThermoFisher HistoStar Embedding Workstation), and cut at a thickness of 5 μm on a microtome (Leica HistoCore Autocut). Tissue sections were mounted on positively charged slides (Fisherbrand Superfrost Plus Microscope Slides) and baked at 60 °C overnight for histology.
Sialic acid lectin staining
To visualize sialic acid modifications in tissue, a unique biotinylated plant-derived lectin from the Sambucas Nigra plant was used to detect the α-2,6 sialic acid bond (Vector Labs Sambucus Nigra Lectin SNA,EBL- Biotinylated B-1305–2). Slides were deparaffinized with serial washes in xylene (3 × , 5 min), 100% ethanol (2 × , 2 min), and 95% ethanol (1 × , 2 min) and flushed in DiH2O (Leica ST5010 Autostainer XL). Heat-induced epitope retrieval was performed by steaming slides in DiH20 for 30 min. After deparaffinization and antigen retrieval, tissue slides were pretreated with Protein Blocking Solution (ThermoFisher SuperBlock™ T20 37,536) for 10 min, 30% hydrogen peroxide (Fisher Chemical H325-100) for 10 min, streptavidin blocking (Vector Labs SP-2002) for 15 min, biotin blocking (Vector Labs SP-2002) for 15 min, and carbo-free blocking solution (Vector Labs SP-5040) for 30 min. Then, SNA lectin was applied to the tissue at the concentration of 1.75 μg/ml (in PBS) for 30 min, followed by incubation of VECTASTAIN Elite ABC peroxidase (Vector Labs PK-6100) for 30 min at room temperature. Lastly, colorization was achieved with 3,3′-diaminobenzidine chromogen (Vector Labs ImmPACT DAB SK-4105) for 5 min. The same protocol was used to visualize α-2,3 sialic acid bond (Vector Labs B-1265) at 2.5 μg/ml (in PBS) for 30 min. Murine kidney tissue was used as a positive control (Supplementary Fig. 2a, b).
Immunohistochemistry (IHC)
Following deparaffinization, beta-amyloid tissue slides were steamed in DiH20 for 30 min and washed in a solution of Tris Buffered Saline with 0.1% Tween 20 detergent. On the Leica ST5010 Autostainer, tissue was quenched in 3% hydrogen peroxide for 10 min and blocked in 2.5% normal goat serum (Sigma G9023) for 15 min. Pan-Aβ rabbit monoclonal antibody D54D2 (1:1000; Cell Signaling) label residues Aβ-37, Aβ-38, Aβ-39, Aβ-40, and Aβ-42 prepared in universal antibody dilution (Sigma U3635) were incubated for 45 min. Following primary antibody incubation, tissue was incubated with rabbit IgG secondary antibody (VisUCyte™ HRP Polymer VC003-025) for 45 min. Colorization was achieved with 3,3-′diaminobenzidine chromogen (BD Bioscience 550,880) for 5 min. All slides were counterstained with Gil 1 Hematoxylin (Epredia 6,765,006) as a nuclear marker, enhanced with Scott’s Tap Bluing H2O (CM4951W). Lastly, slides were dehydrated with serial washes in 95% ethanol (1 × , 2 min), 100% ethanol (2 × , 2 min), and xylene (3 × , 2 min) and coverslipped with permanent xylene-soluble mounting media (Richard-Allan Scientific™ Cytoseal™ XYL).
Immunofluorescence (IF)
Sialic acid detection
IF was performed to visualize α-2,6 sialic acid colocalization with microglia and amyloid plaques. Microglia and amyloid plaques were labeled with a primary antibody targeting ionized calcium binding adaptor protein 1 (IBA1, 1:1000, Wako) and residues 1–4 of human Aβ42 and Aβ40 (MoAB, 1:1000, Millipore), respectively, for 45 min. Combining the aforementioned primary lectin and IHC protocols, Streptavidin DyLight 488 (Vector Labs SA-5488 at 20 μg/ml in PBS), goat anti-rabbit AlexaFluor 555 (1:500), and goat anti-mouse AlexaFluor 647 (1:500) secondary incubations were performed in the dark for 45 min. Autofluorescence artifact was blocked with incubation in Sudan Black B solution (2 min), and slides sealed with Fluoromount-G semi-permanent mounting media containing DAPI counterstain (SouthernBioTech 0100–20).
IHC and IF image acquisition
Brightfield IHC images were acquired and processed using an Olympus LC3 microscope (with U-TV0.63XC camera, 10 × /0.25, 40 × /0.65 FN20 plan objective) and LCmicro 2.2 software. IF images were captured on a Zeiss LSM 880 inverted confocal microscope (LSM 880 Indimo AxioObserver) using a plan Apochromat 40X/1.4 oil DIC M27 oil objective for single planar images and Apochromat 63 × /1.4 oil DIC M27 emersion objective to generate z-stack images, averaging fifteen 0.45-μm interval stacks. Images were processed using ZEN 2.3 (blue edition) software. Whole hemibrain images were captured at 20 × /0.45 PL FL on a BioTek Cytation 5 automated widefield fluorescent microscope and images stitched into montages using the Gen5 3.11.19 software.
IHC and IF analysis
To quantify plaque changes, three measures of plaque coverage were determined using Image J image processing software with Fiji version 1.53q. Color deconvolution was used for total plaque count in the cortical region of interest with the Image J cell counter feature and for plaque ratio of cored-to-diffuse plaques using the nuclear channel to compare cored and non-cored (diffuse) plaques. Plaque burden was determined using Otsu Dark image thresholding to quantify the burden of staining. Analysis of plaque differences based on oseltamivir treatment was calculated with non-parametric T-tests on GraphPad Prism with Standard Deviation (SD). Imported fluorescent images were split into individual channels for thresholding. Percent area coverage of the stains were determined by dividing the total area of stain by the area of the image. Image J analysis macros are available in the Supplementary materials. Two-Way ANOVA (genotype × treatment) with Bonferroni’s correction was used to analyze percent coverage of SA and microglia ± SD. To quantitatively determine the location of α-2,6 SA, a pixel-to-pixel correlation was performed for correlation pairs of SA-to-microglia and SA-to-plaques on Image J. Pearson’s correlation was employed to analyze pixel-to-pixel ratios using above value thresholds only. To present this data, Pearson correlation coefficient r values were used to represent the strength and direction of the relationship between the two variables (SA to microglia) and (SA to plaque). The r values were then compared across genotypes with a 2-way ANOVA (genotype × correlation pairs) with post-hoc Bonferroni’s test. The orthogonal view feature in Image J was used to visualize α-2,6 SA localization.
Results
Visualization of sialic acid in 5XFAD brain tissue
Previous studies have not defined how cell specific sialylation or tissue microenvironment SA change with age and disease. To visualize the distribution of α-2,6 SA in the brain, coronal sections from 5XFAD and WT mice were stained with SNA lectin (brown) and counterstained with hematoxylin (blue) as a nuclear marker (Fig. 1a–c). To compare visual distribution of sialic acid linkage bonds, we compared α-2,6 SA to α-2,3 SA, a bond type also enriched in the brain. The chemical structures are present in Fig. 6a, b. There was a unique distribution of α-2,6 SA in 5XFAD brain tissue (Fig. 1a, b) compared WT (Fig. 1c) that was not appreciable with α-2,3 MAL II lectin staining (Fig. 1d–f). In 5XFAD mice, there was α-2,6 SA signal throughout the brain clustered around the core of Aβ plaques in a manner that resembled plaque associated microglia (Fig. 1a, b). This was an intriguing finding because the Aβ plaques were not labeled with an antibody, yet clearly visible with the lectin staining. While the lectins used to visualize SA patterns identified interesting morphologies, they are not cell type specific. Therefore, further histology was required to validate the localization of α-2,6 SA in the brain architecture to better understand what features of the plaque environment are sialylated.

Observation of sialic acid bond types in 12-month-old 5XFAD and WT brain tissue. Greater α-2,6 sialylation visible compared to α-2,3 sialylation. Tissue samples were stained with bond specific lectins; 10 × images, scale bar = 50 μm; 40 × images, scale bar = 20 μm. a Representative image (10 ×) of α-2,6 sialylation in 5XFAD. b Higher magnification cortical view (40 ×) α-2,6 sialylation surrounding an Aβ plaque. c Representative image (40 ×) of α-2,6 sialylation in WT brain tissue. d Representative image (10 ×) of α-2,3 sialylation in 5XFAD. e Higher magnification cortical view (40 ×) α-2,3 sialylation surrounding an Aβ plaque. f Representative image (40 ×) of α-2,3 sialylation in WT
Behavioral effects of oseltamivir treatment
Mice were treated twice daily for five days with oral oseltamivir in line with clinical standards for patients prescribed anti-viral therapy and allowed for the investigation of acute side effects, with only 2 two days of treatment, and delayed effects after treatment concluded (Fig. 2a). This timeline was important to tease apart acute and delayed side effects because aged patients have reported both classes of neuropsychiatric events. To characterize acute and delayed behavioral changes upon oseltamivir treatment in the context of aging and amyloid pathology, 12-month-old WT and 5XFAD mice were assessed with OF and NOR testing at different time points during drug treatment. On day 3 of drug administration, all mice were assessed in OF to examine acute anxiety-like behavior and retested on day 7 for any prolonged changes. There was not a significant difference in anxiety-like phenotype based on time spent in the center of the arena during OF for drug treatment (Supplementary Fig. 1). A statistically significant difference was observed for velocity between 5XFAD OST (n = 16, F = 9, M = 7), 5XFAD VEH (n = 16, F = 9, M = 7), WT OST (n = 12, F = 6, M = 6), and WT VEH (n = 13, F = 6, M = 7) groups (p = 0.0141), with further interaction of testing day (p = 0.0392; Fig. 2b). Additionally, there was a statistically significant difference for velocity on the first day of OF based on genotype (p = 0.0079) that revealed the 5XFAD group lacked normal habituation behavior to the test environment and did not display a reduction in velocity over time, while the WT did (p = 0.0002; Fig. 2c). Across all evaluations, there was no effect of oseltamivir treatment in OF behavior. To determine if oseltamivir induced any delayed changes in recognition memory, mice were assessed using the NOR paradigm. A statistically significant difference was observed for velocity between 5XFAD OST (n = 15, F = 8, M = 7), 5XFAD VEH (n = 14, F = 9, M = 5), WT OST (n = 9, F = 4, M = 5), and WT VEH (n = 11, F = 4, M = 7) groups (p = 0.0180; Fig. 2d). DI was used to observe changes in recognition memory. There was a statistical difference between 5XFAD OST (n = 15), 5XFAD VEH (n = 14), WT OST (n = 9), and WT VEH (n = 11) groups (p = 0.0335) yet no difference based on oseltamivir treatment (Fig. 2e). Overall, oseltamivir treatment did not affect behavioral parameters as measured by either OF or NOR and substantiates the hyperactivity phenotype in 5XFAD mice.

No behavioral changes with oseltamivir treatment. Treatment of oseltamivir (OST) and ddH20 vehicle (VEH) for 5 days; evaluated with open field (OF) and novel object recognition (NOR). a Graphical description of drug administration and behavioral testing. b OF investigation of treatment effects on locomotion. A 3-way repeated measures ANOVA of velocity comparing drug treatment and genotype across test days (n = 56). Main effect of genotype (p = 0.0141) and interaction of day X genotype (p = 0.0392). c OF habituation attenuation in 5XFAD in day 1 of testing. A 3-way ANOVA of velocity between the first and last minute of OF test demonstrated a main effect of time (p = 0.0079) and interaction of minute x genotype (p = 0.0002). d NOR investigation of treatment effects on learning and memory. A 3-way repeated measures ANOVA of velocity comparing drug treatment and genotype across test days (n = 50, 6 removed for lack of exploration) displayed a main effect of genotype (p = 0.0180). e Discrimination Index calculated from day 2 of NOR. 2-Way ANOVA indicates a main effect of genotype (p = 0.0335). Data is graphed as mean ± SEM
Effects of drug treatment on plaque pathology
Next, we investigated whether treatment with oseltamivir altered Aβ plaque distribution, burden, or morphology by performing immunohistochemical analysis on a subset of 5XFAD mice tested for behavior. Using the anti-Aβ antibody, we quantified representative images of Aβ cored and diffuse plaques in coronal sections focused on the primary somatosensory cortex (Fig. 3a, b, d, e; 40 × Fig. 3c, f; brain region Fig. 3g). There was no significant difference between oseltamivir-treated and vehicle-treated 5XFAD mice with respect to Aβ plaque burden (p = 0.429; Fig. 3h), Aβ plaque count (p = 0.195; Fig. 3i), or the ratio of dense cored and diffuse Aβ plaques (p = 0.999; Fig. 3j).

No changes in plaque pathology. 5XFAD mice treated with OST (n = 5) or ddH2O vehicle (n = 6) were immunostained with anti-Aβ antibody (D54D2) and assessed for plaque pathology changes or morphological differences in the somatosensory subsection of the cerebral cortex; 10 × images, scale bar = 50 μm; 40 × images, scale bar = 20 μm. a–b Representative images (10 ×) of cored and diffuse Aβ plaques from OST treated 5XFAD mice. c Representative image (40 ×) of cored and diffuse Aβ plaques from female OST 5XFAD mice. d–e Representative images (10 ×) of cored and diffuse Aβ plaques from VEH treated 5XFAD mice. f Representative image (40 ×) of cored and diffuse Aβ plaques from female VEH 5XFAD mice. g Primary somatosensory cortex (red star) is the region analyzed, atlas available through Matt Gaidica Brain Atlas. h Plaque burden is the amount of area Aβ plaques occupied in the ROI (p = 0.429). Using image thresholding, values for plaque burden were evaluated with non-significant difference based on treatment. i Total plaque count was manually counted in the cortical ROI with color deconvolution (p = 0.195). Plaque count across treatment groups yielded a non-significant difference. j Cored plaques were defined as dense and compact plaques visible with the nuclear setting of color deconvolution and diffuse plaques were defined as disperse plaques with no visible dense core. Plaque ratio was determined with color deconvolution and is a calculated value comparing the number of cored plaques to diffuse plaques, with no difference in plaque ratio (p = 0.999). Data is graphed as mean ± SD
Localization of sialic acid with immunofluorescence
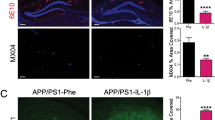
To examine whether oseltamivir treatment mediated changes of SA within the brain, we quantified sialic acid localization with multicolor IF for green, α-2,6 SA linkage (SNA lectin); red, microglia (IBA1); white, Aβ plaques (MoAB); and merged channels. This analysis included a subset of the original cohort of mice, 5XFAD (n = 10) and WT (n = 11), from both treatment groups. Representative images of 5XFAD mice treated with oseltamivir or vehicle display a α-2,6 SA staining pattern that is not affected by treatment (Fig. 4a, b). 5XFAD mice have an overall increased α-2,6 SA coverage compared to WT mice that display minimal SA staining with restrictive blood vessel labeling (Fig. 4c, d). There was statistically different α-2,6 SA area coverage in the cortical ROI between 5XFAD and WT groups (p < 0.001), with no effect of oseltamivir treatment (p = 0.253; Fig. 4e). In addition, there was no difference in microglia area coverage based on genotype (p = 0.208) or treatment (p = 0.067; Fig. 4f).

Increased α-2,6 sialic acid in 5XFAD mice in the plaque microenvironment. Representative images of 5XFAD and WT treated with OST and VEH, immunostained with SNA Lectin for α-2,6 sialic acid (green), IBA1 labeling microglia (red), MoAB demonstrating Aβ plaques (white), and a merged image of the 3 channels; 40 × images, scale bar = 100 μm. a Representative image 5XFAD VEH. b 5XFAD OST. c WT VEH. d WT OST. e A 2-way ANOVA of percent SA coverage in the 40 × ROI displayed a main effect of genotype (p < 0.0001; n = 21). Across groups, OST had significantly more SA coverage (OST p < 0.001; VEH p = 0.0016). f A 2-way ANOVA of percent microglia coverage was non-significant. Percent microglia coverage was defined as the amount of IBA1 positive microglia stain divided by the area of the image multiplied by 100. Data is graphed as mean ± SD
Sialic acid in the plaque microenvironment
Previous studies have not defined the location of SA in the plaque microenvironment. To assess SA localization in the plaque microenvironment, we collected confocal z-stacks and generated orthogonal views to qualitatively understand the relationship between α-2,6 SA, microglia, and Aβ plaques. 5XFAD brains, regardless of treatment, display overlap of SA (green) and microglia (red) channels indicating colocalization of these markers (Fig. 5a, b). In contrast, WT brains did not display a similar channel overlap (Fig. 5c, d). Wildtype sections had low levels of immunofluorescent signal in the Aβ plaques channel that was not positive for Aβ plaques. This was verified with technical controls. To quantitatively evaluate this colocalization, we imaged the SA channel (green) merged with the microglia channel (red) to visualize colocalization, named SA-Microglia (Fig. 5e). Similarly, we imaged the SA channel (green) merged with the Aβ plaque channel (pseudo red) in the same field of view to visualize colocalization, named SA-Plaque (Fig. 5f) for 5XFAD = 6 and WT = 6. Then, we performed Pearson correlation analysis of the image pixel values for the SA-Microglia and SA-Plaque comparisons to determine the localization of SA across genotype. Specifically, the Pearson correlation coefficient (r) was calculated for the SA-Microglia and SA-Plaque comparison for each genotype. After calculating r values, correlation pairs were compared with a 2-way ANOVA, with representative images provided of multicolor IF (Fig. 5e, f). A statistically significant main effect of genotype (5XFAD vs WT) was observed for a decrease in r values for WT in the comparison of SA-Microglia as well as SA-Plaques (p < 0.0001). Notably, post hoc comparisons shed light on the differences between correlation pairs. Within the SA-Microglia correlation, 5XFAD had significantly greater r values compared to WT (p < 0.0001); the same effect was observed for the SA-Plaque correlation (p < 0.0001). Importantly, within the 5XFAD genotype, the SA-Microglia r value (mean r = 0.522) was significantly greater (p < 0.0001) to the 5XFAD SA-Plaque r value (mean r = 0.262; Fig. 5g). These correlations quantify our observation that SA is increased on microglia in 5XFAD mice more than on Aβ plaques.

Colocalization and sialylation of microglia in 5XFAD. 5XFAD and WT cortical tissue tripled stained, α-2,6 sialic acid (green), IBA1 labeling microglia (red), and MoAB labeling plaques (grey), merged and visualized with (63 ×) orthogonal view. a 5XFAD OST, b 5XFAD VEH, c WT OST, d WT VEH. e Representative image (40 ×) of the green sialic acid channel merged with red microglia channel to visualize colocalization (in yellow); described as SA-Microglia. f Representative image (40 ×) of the green sialic acid channel merged with Aβ plaque (pseudo red) channel to visualize colocalization, described as SA-Plaque; these individual merged images allow for the calculation of Pearson correlation coefficient r values for the degree of colocalization. g A 2-way ANOVA of above value Pearson correlation coefficient r values for SA-Microglia versus SA-Aβ plaques (n = 12). Data is graphed as mean ± SD
Discussion
The goal of the current study was to determine if oseltamivir could mediate neuropsychiatric side effects via reorganization of SA in the brain, putatively by blocking desialylation at α-2,6 SA bonds via inhibition of Neu1. To develop a clear understanding of sialylation distribution patterns, we developed histological techniques to localize SA in the brain. Our methods build upon previous studies which were unable to resolve spatial distribution of SA in brain in aging or AD. Initially, we observed an interesting pattern of α-2,6 SA that was distinct in 5XFAD mice compared to WT littermates (Fig. 1a, c). In contrast, we did not observe the same staining pattern for α-2,3 SA (Fig. 1d, f), suggesting that reorganization of SA during amyloid pathology is specific to α-2,6. Here, we suggest that α-2,6 SA and α-2,3 SA have different steric shapes, described in Fig. 6, which will differentially bind lectins. With this finding, the biological relevance to continue investigating α-2,6 SA aligns with the strength of visualization in tissue and its role as an immune modulator for communication with other cells [22].

Graphical representation of α-2,6 sialic acid and α-2,3 sialic acid chemical structure and association with microglia and Aβ plaques. Visual representation of sialic acid bond specific structures and our theory of sialylated microglia interaction with amyloid pathology. a Chemical structure of α-2,6 sialic acid linkage bond, created with ChemSpider software. b Chemical structure of α-2,3 sialic acid linkage bond. c Graphical depiction of a sialylated microglia with a magnified view of the bond specific attachments of α-2,6 and α-2,3 sialic acid modifications. The scissor icon represents Neu1 enzyme and the cleavage of α-2,6 sialic acid. The prescription bottle represents oseltamivir and the inhibitory effects on Neu1. The right side of the figure represents an α-2,6 sialylated microglia surrounding a beta-amyloid plaque. This graphic was created with Biorender software
Upon quantification, we found 5XFAD mice displayed significantly greater α-2,6 SA compared to WT littermates (Fig. 4e). This aligns with the previous studies showing that SA levels differ between disease states. Patients with AD have significantly higher levels of serum SA compared to controls [28] suggesting that SA levels may influence Aβ deposition. Further, enzymes that transfer SA residues onto terminal glycoconjugates decline in AD, while recycling of SA residues via specific neuraminidase enzymes is vital for recognition by specific Siglec receptors [27]. Reports suggest that CSF metabolomics revealing patients with MCI have higher SA metabolism compared to normal controls, suggesting glycosylation changes precede clinical progression to AD [15]. Others have shown that levels of brain sialylation decrease in patients with MCI or AD but increase in the blood [27, 35]. These studies demonstrate variations in brain, blood, and CSF levels of sialylation which require further investigation, but still lack a clear representation of the spatial organization of SA in AD.
While it is unclear how reorganization of SA during aging and MCI contributes the pathogenesis and progression of AD, our data suggests a role for α-2,6 SA in the plaque microenvironment and that α-2,6 SA is localized to plaque-associated microglia and, to a lesser extent, amyloid plaques. We see no significant increases in IBA1 area fraction, suggesting this increase in α-2,6 SA is not simply due to increases in microglia (Fig. 4f). This is apparent in orthogonal views of the plaque microenvironment in 5XFAD mice as well as by Pearson pixel correlation ratios comparing α-2,6 SA, microglia, and Aβ plaques. We demonstrate that there is significant overlap of the α-2,6 SA and microglia in 5XFAD mice (Fig. 5a, b) and α-2,6 SA is more positively correlated with microglia compared to Aβ plaques within the 5XFAD mice (Fig. 5e, f, g). These data suggest that microglia are more sialylated in the 5XFAD mouse model compared to Aβ plaques.
We posit that α-2,6 SA modification of microglia may contribute to alterations in microglia function during the pathogenesis and progression of AD. SA residues on the cell surface facilitate cellular interaction and immune defense responses [49]. Intriguingly, cell surface sialylation is used as a self-recognition system to prevent immune response or detection by other immune cells [24]. Microglia use this SA recognition system in collaboration with receptors to modulate activity. Indeed, inhibitory Siglec receptors that bind SA residues as ligands block microglia activation [26]. It is important to understand α-2,6 SA and α-2,3 SA serve as signaling molecules for cell–cell interactions, cell migration, and cell adhesion [41]. Previous studies have described the role of sialylation on microglia as a mechanism of immune inhibition [2]. Sialylation of microglia inhibits the inflammatory activation of microglia by inducing inhibitory receptors Siglec 2 or Siglec 3 to reduce microglial phagocytosis [10, 43]. Importantly, α-2,6 SA serves as a preferred ligand for the inhibitory receptor Siglec 2 which has increased levels on aged microglia and negatively regulates microglia phagocytosis [1, 37]. Notably, the isoform that lacks exon 2 in the inhibitory SA receptor Siglec 3 has been identified as a risk factor for AD with increased expression in AD patients [50]. As α-2,6 SA is a ligand for this receptor, this SA modification may play some role in resilience or risk of AD [24, 32]. The changes in Siglec expression in aging and disease solidify the importance of understanding SA levels and signaling in vivo.
Importantly, desialylation relieves immune inhibition and allows microglial activation and/or phagocytosis to occur [52]. A study focused on the immune response in B cells suggests that Neu1 preferentially acts on glycoproteins with some impact on ganglioside sialylation [45]. In our study, we take this information from B cells which share similar sialylation mechanisms to microglia and predict that Neu1 is likely acting on glycoproteins with some involvement of gangliosides. In our study, inhibition of Neu1 with oseltamivir was predicted to decrease SA cleavage on glycoproteins. To support this, it would be useful to compare sialylation of glycoproteins and glycolipids in 5XFAD mice treated with oseltamivir. Regarding gangliosides, work done by Crain and Shen demonstrated oseltamivir treatment blocks morphine’s hyperalgesic effects by decreasing neuronal levels of monosialotetrahexosylganglioside or GM1 [8]. Given this finding, we believe we are looking at sialylation of microglia and oseltamivir may not be impacting GM1 levels as significantly as this study reported in neurons. Investigating gangliosides in microglia, Galleguillos and colleagues suggest GM1 levels decrease microglia inflammatory response in the presence of SA modifications on GM1 glycan headgroup and lipid tail [13]. Similar to glycoproteins, gangliosides play a role in immunomodulation, cell communication, and cell signaling in the brain [38]. These are very intriguing results on the relevance of gangliosides; we believe that future studies investigating the role of sialylation on glycosphingolipids and glycoproteins would be invaluable to develop a better understanding of the influence of sialylation in the context of different brain pathologies.
In order to fully understand the role of neuraminidase function, previous studies suggest knocking out Neu1 can change behavior and brain level sialic acid patterns for α-2,6 SA in zebrafish [18]. However, we saw no effect of oseltamivir treatment on microglia labeling with IBA1 or α-2,6 SA. The 5XFAD mouse model develops robust Aβ plaque deposition and recapitulates many neuroinflammatory phenotypes of human AD [34],therefore, it would be of interest to test whether manipulation of SA via mechanisms different from Neu1 inhibition with oseltamivir would alter the neuroinflammatory phenotype of this model. Spatial and cell type specific analyses of components of the SA pathway throughout the lifespan could reveal glycosylation patterns that mediate the pathogenesis of age-associated neurodegenerative diseases.
While we found intriguing differences in α-2,6 SA between WT and 5XFAD mice, we did not find a significant effect of oseltamivir treatment in any of the parameters evaluated. Despite data from other studies suggesting an effect of oseltamivir on behavior in humans [6, 14] and in rodents with other stimulants [20], 5-day oral treatment of oseltamivir had no effect on OF or NOR behavior (Fig. 2). While a longer treatment may have resulted in stronger effects, the decision to use a 5-day treatment was two-fold. First, an acute 5-day treatment of oseltamivir is in alignment with clinical dosing guidelines for patients with influenza [19]. Secondly, prolonged or chronic treatment has been associated with liver toxicity and oxidative damage in rats [11]. Therefore, an acute treatment paradigm was most appropriate and translationally relevant. Further, oseltamivir can cross the blood brain barrier [30] and alter mammalian tissue sialylation levels [9] providing support for the potential alteration of brain sialylation levels by oseltamivir, although we did not observe this.
To date, the exact mechanism of increased sialylation is undetermined in the 5XFAD mouse model. A general working theory is through the amyloidogenic pathway to process Aβ, the enzyme beta-site APP cleaving enzyme (BACE1) may be colocalized with the enzyme ST6Gal-1 which would increase the levels of sialic acid present, creating an increase in APP sialylation [54]. While this has not been tested in the 5XFAD mouse model, this model harbors transgenes for the development of amyloid pathology. In our study, we show that the increased sialylation is most abundant on the microglia cell surface. We posit that both the increase in SA on plaque-associated microglia in 5XFAD mice and the lack of effect of oseltamivir on SA levels in our study could be due to reduced Neu1 enzyme activity or abundance in 5XFAD mice. Therefore, we examined publicly available RNA sequencing datasets on aging 5XFAD brain and found that 8-month-old 5XFAD mice have significantly decreased Neu1 expression compared to age-matched WT mice [12]. Additionally, 5XFAD mice have a robust age-related increase in the expression of sialyltransferase ST6Gal1, responsible for α-2,6 SA, in both the cortex and hippocampus compared to WT [12]. As the abundance and activity of upstream sialyltransferase enzymes change with age in this model, this may account for the increase in α-2,6 SA we observe in 5XFAD mice. We postulate the activated microglia in 5XFAD mice are reacting toward the amyloid pathology by utilizing the immune response of sialylation and subsequent receptors. Thus, this increase in sialylation may be a signal of continued disease progression in our 12-month-old mouse cohort.
In alignment with previously published data, we observed that 5XFAD mice have consistently higher velocity compared to WT (Fig. 2b, d) and lack habituation to the testing environment (Fig. 2c) which is consistent with previous literature demonstrating that 5XFAD mice are hyperactive [34]. We did not observe any differences in plaque count, deposition, or morphology following oseltamivir treatment (Fig. 3), yet there was a visual decrease in male OST (Fig. 3a) compared to male VEH (Fig. 3d) which is likely due to biological variability rather than treatment effects. While the 5-day PO BID oseltamivir regimen is similar to prescription guidelines for patients [44] and shows antiviral efficacy in mice [53], a limitation of our study is the absence of influenza infection. It is well established that infection and acute illness are known to induce delirium in aged patients and patients with AD [5]. While there are many predisposing factors associated with age-related delirium, the lack of viral infection could be a major factor in the neuropsychiatric effects of oseltamivir observed in human patients that we are missing in this study design. Further, mice in this study were aged only to 12 months which may not be sufficient to capture any age-dependent interactions between oseltamivir and amyloid pathology. While this study investigated middle aged mice and the impact of Neu1 inhibition, it would be advantageous to use aged mice, 18 months and older, to probe pathogenic Aβ interaction with SA residues. Additionally, oseltamivir is also known to induce neuropsychiatric side effects in young people. In Japan, a cohort of patients 16 years or younger reported adverse behavioral side effects [6], suggesting that viral infection may be a driving cause of the adverse side effects. While it is difficult to distinguish viral infection, treatment, and underlying pathology as the causal driver for these reports, this study aimed to clarify the localization of SA under drug administration in this mouse model of amyloid pathology.
Conclusion
Previous reports have highlighted changes in glycosylation patterns in aging and AD along with increased levels α-2,6 SA. However, prior to the present study, very little was known about the spatial distribution of α-2,6 SA relative to amyloid pathology. Our findings suggest that plaque-associated microglia are highly sialylated and are resistant to to oseltamivir. We posit that increased levels of SA may be due to increased expression of the sialyltransferase ST6Gal1, while resistance to oseltamivir may be mediated by age- and disease-associated decreases in Neu1 expression. Overall, the role of SA in plaque associated microglia warrants further investigation due to the potential role of SA in interfering with microglia immune recognition of and response to amyloid pathology via Siglec receptors.
Data Availability
The datasets generated during the current study are available from the corresponding author on reasonable request.
Abbreviations
- Neu1:
-
neuraminidase 1
- AD:
-
Alzheimer’s Disease
- 5XFAD:
-
Five familiar Alzheimer’s disease mutations
- Aβ:
-
amyloid beta
- PTM:
-
post-translational modification
- SA:
-
sialic acid
- Siglec:
-
sialic acid binding immunoglobulin-like lectin
- CSF:
-
cerebral spinal fluid
- MCI:
-
mild cognitive impairment
- MS:
-
mass spectrometry
- APP:
-
amyloid precursor protein
- WT:
-
wild-type
- PCR:
-
polymerase chain reaction
- OF:
-
open field
- NOR:
-
novel object recognition
- DI:
-
discrimination index
- IHC:
-
immunohistochemistry
- IF:
-
immunofluorescence
- ROI:
-
region of interest
References
Aires V, Coulon-Bainier C, Pavlovic A, Ebeling M, Schmucki R, Schweitzer C, Kueng E, Gutbier S, Harde E. CD22 blockage restores age-related impairments of microglia surveillance capacity. Front Immunol. 2021;0:2096.
Allendorf DH, Puigdellívol M, Brown GC. Activated microglia desialylate their surface, stimulating complement receptor 3-mediated phagocytosis of neurons. Glia. 2020;68:989–98.
Annunziata I, Patterson A, Helton D, Hu H, Moshiach S, Gomero E, Nixon R, D’Azzo A. Lysosomal NEU1 deficiency affects amyloid precursor protein levels and amyloid-β secretion via deregulated lysosomal exocytosis. Nat Commun. 2013;4:2734.
Bhide GP, Colley KJ. Sialylation of N-glycans: mechanism, cellular compartmentalization and function. Histochem Cell Biol. 2017;147:149–74.
Bowman K, Jones L, Masoli J, Mujica-Mota R, Strain D, Butchart J, Valderas JM, Fortinsky RH, Melzer D, Delgado J. Predicting incident delirium diagnoses using data from primary-care electronic health records. Age Ageing. 2020;49:374–381. Available at: https://academic.oup.com/ageing/article/49/3/374/5814887. Accessed 26 Aug 2022.
Chen R, Fang Z, Huang Y. Neuropsychiatric events in an adult patient with influenza a (H3N2) treated with oseltamivir (Tamiflu): A case report. BMC Infect Dis. 2019;19:4–7.
Cowan CB, Patel DA, Good TA. Exploring the mechanism of β-amyloid toxicity attenuation by multivalent sialic acid polymers through the use of mathematical models. J Theor Biol. 2009;258:189–97.
Crain SM, Shen KF. Neuraminidase inhibitor, oseltamivir blocks GM1 ganglioside-regulated excitatory opioid receptor-mediated hyperalgesia, enhances opioid analgesia and attenuates tolerance in mice. Brain Res. 2004;995:260–266. Available at: https://pubmed.ncbi.nlm.nih.gov/14672816/. Accessed 14 Oct 2022.
De Oliveira JT, Santos AL, Gomes C, Barros R, Ribeiro C, Mendes N, De Matos AJ, Vasconcelos MH, Oliveira MJ, Reis CA, Gärtner F. Anti-influenza neuraminidase inhibitor oseltamivir phosphate induces canine mammary cancer cell aggressiveness. PLoS One. 2015;10:e0121590. Available at: https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0121590. Accessed 19 Oct 2022.
Duan S, Paulson JC. Siglecs as immune cell checkpoints in disease. Annu Rev Immunol. 2020;38:365–395. Available at: https://pubmed.ncbi.nlm.nih.gov/31986070/. Accessed 26 Aug 2022.
El-Sayed WM, Al-Kahtani MA. Potential adverse effects of oseltamivir in rats: males are more vulnerable than females. https://doi.org/10.1139/y11-060 2011;89:623–630. Available at: https://cdnsciencepub.com/doi/10.1139/y11-060. Accessed 14 Oct 2022.
Forner S, et al. Systematic phenotyping and characterization of the 5xFAD mouse model of Alzheimer’s disease. Sci Data. 2021;8(1):1–16 Available at: https://www.nature.com/articles/s41597-021-01054-y. Accessed 19 Oct 2022.
Galleguillos D, Wang Q, Steinberg N, Zaidi A, Shrivastava G, Dhami K, Daskhan GC, Schmidt EN, Dworsky-Fried Z, Giuliani F, Churchward M, Power C, Todd K, Taylor A, Macauley MS, Sipione S. Anti-inflammatory role of GM1 and other gangliosides on microglia. J Neuroinflammation. 2022;19. Available at: https://pmc/articles/PMC8739653/. Accessed 16 Feb 2023.
Gupta YK, Meenu M, Mohan P. The Tamiflu fiasco and lessons learnt. Indian J Pharmacol. 2015;47:11–6.
Hajjar I, Liu C, Jones DP, Uppal K. Untargeted metabolomics reveal dysregulations in sugar, methionine, and tyrosine pathways in the prodromal state of AD. 2020. Available at: https://doi.org/10.1002/dad2.12064. Accessed 10 Sept 2021.
Hama R. The mechanisms of delayed onset type adverse reactions to oseltamivir. Infect Dis (London, England). 2016;48:651. Available at: https://pmc/articles/PMC4973146/. Accessed 5 Dec 2021.
Hatori A, Arai T, Yanamoto K, Yamasaki T, Kawamura K, Yui J, Konno F, Nakao R, Suzuki K, Zhang MR. Biodistribution and metabolism of the anti-influenza drug [11C]oseltamivir and its active metabolite [11C]Ro 64–0802 in mice. Nucl Med Biol. 2009;36:47–55.
Ikeda A, Komamizu M, Hayashi A, Yamasaki C, Okada K, Kawabe M, Komatsu M, Shiozaki K. Neu1 deficiency induces abnormal emotional behavior in zebrafish. Sci Rep. 2021;11(1):1–15 Available at: https://www.nature.com/articles/s41598-021-92778-9. Accessed 19 Oct 2022.
Ison MG, Portsmouth S, Yoshida Y, Shishido T, Mitchener M, Tsuchiya K, Uehara T, Hayden FG. Early treatment with baloxavir marboxil in high-risk adolescent and adult outpatients with uncomplicated influenza (CAPSTONE-2): a randomised, placebo-controlled, phase 3 trial. Lancet Infect Dis. 2020;20:1204–1214. Available at: http://www.thelancet.com/article/S1473309920300049/fulltext. Accessed 25 Aug 2022.
Izumi Y, Tokuda K, O’Dell KA, Zorumski CF, Narahashi T. Synaptic and behavioral interactions of oseltamivir (Tamiflu) with neurostimulants. Hum Exp Toxicol. 2008;27:911–917. Available at: https://pubmed.ncbi.nlm.nih.gov/19273546/. Accessed 12 Dec 2021.
Klaus C, Hansen JN, Ginolhac A, Gérard D, Gnanapragassam VS, Horstkorte R, Rossdam C, Buettner FFR, Sauter T, Sinkkonen L, Neumann H, Linnartz-Gerlach B. Reduced sialylation triggers homeostatic synapse and neuronal loss in middle-aged mice. Neurobiol Aging. 2020;88:91–107. Available at: https://doi.org/10.1016/j.neurobiolaging.2020.01.008.
Klaus C, Liao H, Allendorf DH, Brown GC, Neumann H. Sialylation acts as a checkpoint for innate immune responses in the central nervous system. Glia. 2021;69:1619–36.
Li F, Ding J. Sialylation is involved in cell fate decision during development, reprogramming and cancer progression. Protein Cell. 2019;10:550–565. Available at: https://doi.org/10.1007/s13238-018-0597-5.
Liao H, Klaus C, Neumann H. Control of innate immunity by sialic acids in the nervous tissue. Int J Mol Sci. 2020;21:5494. Available at: https://www.mdpi.com/1422-0067/21/15/5494/htm. Accessed 22 Aug 2022.
Liao H, Winkler J, Wißfeld J, Shahraz A, Klaus C, Neumann H. Low molecular weight polysialic acid prevents lipopolysaccharide-induced inflammatory dopaminergic neurodegeneration in humanized SIGLEC11 transgenic mice. Glia. 2021. Available at: https://onlinelibrary.wiley.com/doi/full/10.1002/glia.24073. Accessed 20 Aug 2021.
Linnartz-Gerlach B, Kopatz J, Neumann H. Siglec functions of microglia. Glycobiology. 2014;24:794–799. Available at: https://pubmed.ncbi.nlm.nih.gov/24833613/. Accessed 20 Aug 2021.
Maguire TM, Thakore J, Dinan TG, Hopwood S, Breen KC. Plasma sialyltransferase levels in psychiatric disorders as a possible indicator of HPA axis function. Biol Psychiatry. 1997;41:1131–6.
Marino JH, Tan C, Davis B, Han ES, Hickey M, Naukam R, Taylor A, Miller KS, Van De Wiele CJ, Teague TK. Disruption of thymopoiesis in ST6Gal I-deficient mice. Glycobiology. 2008;18:719–26.
McElhaney JE, Verschoor CP, Andrew MK, Haynes L, Kuchel GA, Pawelec G. The immune response to influenza in older humans: beyond immune senescence. Immun Ageing. 2020;17. Available at: https://pmc/articles/PMC7204009/. Accessed 18 July 2022.
Morimoto K, Nakakariya M, Shirasaka Y, Kakinuma C, Fujita T, Tamai I, Ogihara T. Oseltamivir (Tamiflu) efflux transport at the blood-brain barrier via P-glycoprotein. Drug Metab Dispos. 2008;36:6–9. Available at: https://dmd.aspetjournals.org/content/36/1/6. Accessed 14 Oct 2022.
Nagamine S, Yamazaki T, Makioka K, Fujita Y, Ikeda M, Takatama M, Okamoto K, Yokoo H, Ikeda Y. Hypersialylation is a common feature of neurofibrillary tangles and granulovacuolar degenerations in Alzheimer’s disease and tauopathy brains. Neuropathology. 2016;36:333–345. Available at: https://onlinelibrary.wiley.com/doi/full/10.1111/neup.12277. Accessed 19 Aug 2022.
Naj AC, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;435 43:436–441. Available at: https://www.nature.com/articles/ng.801. Accessed 12 Jan 2023.
Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–10140. Available at: https://pubmed.ncbi.nlm.nih.gov/17021169/. Accessed 26 Aug 2022.
Oblak AL, et al. Comprehensive evaluation of the 5XFAD mouse model for preclinical testing applications: a MODEL-AD study. Front Aging Neurosci. 2021;13. Available at: https://pmc/articles/PMC8346252/. Accessed 18 Apr 2022.
Palmigiano A, Barone R, Sturiale L, Sanfilippo C, Bua RO, Romeo DA, Messina A, Capuana ML, Maci T, Le Pira F, Zappia M, Garozzo D. CSF N-glycoproteomics for early diagnosis in Alzheimer’s disease. J Proteomics. 2016;131:29–37.
Peng W, Paulson JC. CD22 ligands on a natural N-glycan scaffold efficiently deliver toxins to B-lymphoma cells. J Am Chem Soc. 2017;139:12450–8.
Pluvinage JV, Haney MS, Smith BAH, Sun J, Iram T, Bonanno L, Li L, Lee DP, Morgens DW, Yang AC, Shuken SR, Gate D, Scott M, Khatri P, Luo J, Bertozzi CR, Bassik MC, Wyss-Coray T. CD22 blockade restores homeostatic microglial phagocytosis in ageing brains. Nat. 2019;5687751 568:187–192. Available at: https://www.nature.com/articles/s41586-019-1088-4. Accessed 16 Aug 2021.
Posse de Chaves E, Sipione S. Sphingolipids and gangliosides of the nervous system in membrane function and dysfunction. FEBS Lett. 2010;584:1748–59.
Pshezhetsky AV, Ashmarina M. Keeping it trim: roles of neuraminidases in CNS function. Glycoconj J. 2018;35:375. Available at: https://pmc/articles/PMC6182584/. Accessed 15 Apr 2022.
Rawal P, Zhao L. Sialometabolism in brain health and Alzheimer’sd. Front Neurosci. 2021;15:1–19.
Schnaar RL, Gerardy-Schahn R, Hildebrandt H. Sialic acids in the brain: gangliosides and polysialic acid in nervous system development, stability, disease, and regeneration. Physiol Rev. 2014;94:461–518.
Siddiqui SS, Springer SA, Verhagen A, Sundaramurthy V, Alisson-Silva F, Jiang W, Ghosh P, Varki A. The Alzheimer’s disease-protective CD33 splice variant mediates adaptive loss of function via diversion to an intracellular pool. J Biol Chem. 2017;292:15312–15320. Available at: https://pubmed.ncbi.nlm.nih.gov/28747436/. Accessed 31 Aug 2021.
Siddiqui SS, Matar R, Merheb M, Hodeify R, Vazhappilly CG, Marton J, Shamsuddin SA, Zouabi HA. Siglecs in brain function and neurological disorders. Cells. 2019;8. Available at: https://pubmed.ncbi.nlm.nih.gov/31546700/. Accessed 13 Aug 2021.
Sur M, Lopez MJ, Baker MB. Oseltamivir. Kucers Use Antibiot A Clin Rev Antibacterial, Antifung Antiparasit Antivir Drugs, Seventh Ed:4580–4609. 2022. Available at: https://www.ncbi.nlm.nih.gov/books/NBK539909/. Accessed 26 Aug 2022.
Ton Tran HT, Li C, Chakraberty R, Cairo CW. NEU1 and NEU3 enzymes alter CD22 organization on B cells. Biophys Rep. 2022;2(3):100064.
Toovey S, Rayner C, Prinssen E, Chu T, Donner B, Thakrar B, Dutkowski R, Hoffmann G, Breidenbach A, Lindemann L, Carey E, Boak L, Gieschke R, Sacks S, Solsky J, Small I, Reddy D. Assessment of neuropsychiatric adverse events in influenza patients treated with oseltamivir. Drug Saf. 2012;2008 3112 31:1097–1114. Available at: https://springerlink.bibliotecabuap.elogim.com/article/10.2165/0002018-200831120-00006. Accessed 18 July 2022.
Varki A. Sialic acids in human health and disease. Trends Mol Med. 2008;14:351. Available at: https://pmc/articles/PMC2553044/. Accessed 13 Aug 2021.
Varki NM, Varki A. Diversity in cell surface sialic acid presentations: implications for biology and disease. Lab Investig. 2007;879 87:851–857. Available at: https://www.nature.com/articles/3700656. Accessed 13 Aug 2021.
Walker JA, Smith KGC. CD22: an inhibitory enigma. Immunology. 2008;123:314. Available at: https://pmc/articles/PMC2433339/. Accessed 10 Sept 2021.
Walker DG, Whetzel AM, Serrano G, Sue LI, Beach TG, Lue LF. Association of CD33 polymorphism rs3865444 with Alzheimer’s disease pathology and CD33 expression in human cerebral cortex. Neurobiol Aging. 2015;36:571–82.
Ward P, Small I, Smith J, Suter P, Dutkowski R. Oseltamivir (Tamiflu®) and its potential for use in the event of an influenza pandemic. J Antimicrob Chemother. 2005;55:i5–i21. Available at: https://academic.oup.com/jac/article/55/suppl_1/i5/2473509. Accessed 6 Jan 2023.
Wei M, Wang PG. Desialylation in physiological and pathological processes: new target for diagnostic and therapeutic development. Prog Mol Biol Transl Sci. 2019;162:25–57.
Wong ZX, Jones JE, Anderson GP, Gualano RC. Oseltamivir treatment of mice before or after mild influenza infection reduced cellular and cytokine inflammation in the lung. Influenza Other Respi Viruses. 2011;5:343. Available at: https://pmc/articles/PMC4942046/. Accessed 26 Aug 2022.
Yang K, Yang Z, Chen X, Li W. The significance of sialylation on the pathogenesis of Alzheimer’s disease. Brain Res Bull. 2021;173:116–123. Available at: https://doi.org/10.1016/j.brainresbull.2021.05.009.
Acknowledgements
The authors would like to thank Mallory Maybrier for histological expertise and Kathleen Bensen for cell counting.
Funding
This work was supported by The National Institutes of Health [T32AG021890 to CF, NS082145 to KFB, R21AG072423 and pilot funding under P30AG013319 to SCH, and P30AG066546 to KFB], the Texas Alzheimer’s Research and Care Consortium to KFB, and the Bartell and Mollie Zachry Endowment for Alzheimer’s Research and Patient Care to KFB.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declre no competing interest.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.

Supplemental Fig. 1
(PNG 196 kb)

Supplemental Fig. 2
(PNG 3507 kb)
Supplement 1
(TXT 2 kb)
About this article

{kind=link}
{kind=link}
Cite this article
Fastenau, C., Wickline, J.L., Smith, S. et al. Increased α-2,6 sialic acid on microglia in amyloid pathology is resistant to oseltamivir. GeroScience 45, 1539–1555 (2023). https://doi.org/10.1007/s11357-023-00761-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11357-023-00761-1