
Abstract
Sepsis-associated encephalopathy (SAE) induces neuroinflammation, which is associated with cognitive impairment (CI). CI is also correlated with aging. We used contrast-enhanced magnetic resonance imaging (MRI), perfusion MRI, and MR spectroscopy to assess long-term alterations in BBB permeability, microvascularity, and metabolism, respectively, in a rat lipopolysaccharide-induced SAE model. Free radical-targeted molecular MRI was used to detect brain radical levels at 24 h and 1 week post-LPS injection. CE-MRI showed increased Gd-DTPA uptake in LPS rat brains at 24 h in cerebral cortex, hippocampus, thalamus, and perirhinal cortex regions. Increased MRI signal intensities were observed in LPS rat brains in cerebral cortex, perirhinal cortex, and hippocampus regions 1 week post-LPS. Long-term BBB dysfunction was detected in the cerebral cortex at 6 weeks post-LPS. Increased relative cerebral blood flow (rCBF) in cortex and thalamus regions at 24 h, decreased cortical and hippocampal rCBF at 6 weeks, decreased cortical rCBF at 3 and 12 weeks, and increased thalamus rCBF at 6 weeks post-LPS, were detected. MRS indicated that LPS-exposed rat brains had decreased: NAA/Cho metabolite ratios at 1, 3, 6, and 12 weeks; Cr/Cho at 1, 3, and 12 weeks; and Myo-Ins/Cho at 1, 3, and 6 weeks post-LPS. Free radical imaging detected increased radical levels in LPS rat brains at 24 h and 1 week post-LPS. LPS-exposed rats were compared to saline-treated controls. We clearly demonstrated BBB dysfunction, impaired vascularity, and decreased brain metabolites, as measures of long-term neuroinflammatory indicators, as well as increased free radicals in a LPS-induced rat SAE model.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Sepsis is a severe acute systemic inflammatory reaction to circulating bacterial toxins. It is a serious and potentially fatal illness and can result in multi-organ dysfunction and failure. Sepsis is associated with both acute and chronic brain dysfunctions (Ning et al. 2017; Taccone et al. 2013). Sepsis-associated encephalopathy (SAE) is a common acute complication of sepsis that is associated with a poor prognosis and cognitive impairment (Jain et al. 2002; Ziaja 2013; Wu et al. 2015; Sun et al. 2015). In general, neuroinflammation is considered a risk factor for cognitive impairment (Sun et al. 2015; Yamanaka et al. 2017). It is also well known that cognitive impairment is a direct outcome of aging (Bettio et al. 2017; Toth et al. 2017; Oedekoven et al. 2015). Clinical and preclinical data has implicated that systemic inflammation can induce long-lasting cognitive complications, particularly in the aged and vulnerable brain (Murray et al. 2012; Sun et al. 2015; Cunningham and Hennessy 2015; Yamanaka et al. 2017). In humans, chronic neurocognitive compromise is well recognized in sepsis survivors, affecting 70% of patients at hospital discharge and 45% at 1 year (Mikkelsen et al. 2012).
SAE is a common complication that subsequently occurs with poor prognosis in septic patients (Ziaja 2013; Bozza et al. 2013). SAE is also associated with cognitive impairment (Ziaja 2013; Wu et al. 2015). Sepsis in a mouse model (cecal ligation/perforation, CLP) resulted in hippocampal-dependent cognitive impairment, as measured in behavioral tests, showing significantly increased distance spent in an open field test, and decreased freezing time to context in a fear conditioning test (Gao et al. 2017). In another study using the CLP sepsis-induced mouse model, long-term cognitive impairment, measured by behavioral outcomes from an inhibitory avoidance test and a Morris water maze, was detected 15 days after septic induction (Hernandes et al. 2014). It was also found that the CD40-CD40 ligand pathway plays a major role in acute neuroinflammation and contributes to long-term cognitive dysfunction following sepsis (Michels et al. 2015).
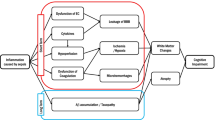
SAE pathophysiology is still poorly understood, but a number of mechanisms-of-actions (MOAs) have been proposed, including mitochondrial and vascular dysfunction, oxidative damage, neurotransmission disturbances, inflammation, and cellular death (Ziaja 2013; Bozza et al. 2013; Berg et al. 2011; Jacob et al. 2011). Oxidative stress is a central MOA of acute brain damage (Bozza et al. 2013). Systemic inflammation induces mitochondrial dysfunction, which is involved in both apoptotic and necrotic cell death pathways, and increased glucose uptake by brain tissues. This results in the diversion of glucose to the pentose phosphate pathway that may contribute to oxidative stress by producing excessive superoxide radicals via NADPH oxidase (Bozza et al. 2013; Berg et al. 2011). In addition, microglia activation results in the secretion of nitric oxide, reactive oxygen species (ROS) and matrix metalloproteinases (MMPs) that can all contribute to blood-brain barrier (BBB) and neuronal damage (Wu et al. 2015). Regarding brain dysfunction in sepsis, it is thought that reactive oxygen and nitrogen species (RONS), generated during a systemic inflammatory response, triggers lipid peroxidation due to a decreased antioxidant activity (Berg et al. 2011). Free radical-induced structural membrane damage also induces neuroinflammation (Berg et al. 2011). The formation of excessive superoxide radicals also depletes ambient nitric oxide in the cerebrovascular bed (Berg et al. 2011). This results in the formation of peroxynitrite, which irreversibly inhibits the mitochondrial electron transport chain, resulting in an increase in mitochondrial release of free radicals, and leads to mitochondrial dysfunction and neuronal bioenergetics failure (Berg et al. 2011). Additionally, free radicals trigger apoptosis via altering intracellular calcium homeostasis in brain regions such as the cerebral cortex and hippocampus, further exacerbating local inflammatory responses further (Berg et al. 2011).
There are a variety of MRI (magnetic resonance imaging) methods that can be used to assess neuroinflammation. Contrast-enhanced MRI (CE-MRI) or dynamic contrast-enhanced (DCE) MRI can be used to assess whether there is increased blood-brain barrier (BBB) permeability, by assessing the uptake of a MRI contrast agent, Gd-DTPA (gadolinium diethylene triamino penta acetic acid), which increases MRI signal intensity (SI), in the brain tissues. Perfusion MR imaging (pMRI) (either arterial spin labeling (ASL), or DCE-MRI) can also be used to assess tissue vascularlity by measuring relative cerebral blood flow (rCBF). Metabolite levels assessed by magnetic resonance spectroscopy (MRS) can also be used as markers of neuroinflammation in neurodegenerative diseases. Our group also developed a free radical-targeted molecular MRI (mMRI) approach to assess in vivo free radicals associated with oxidative stress-related neuroinflammatory diseases, including SAE (Towner et al. 2013a), gliomas (Towner et al. 2013b), and amyotrophic lateral sclerosis (ALS) (Towner et al. 2013c).
It is well established that RONS lead to structural and functional modifications of cellular proteins and lipids, resulting in cellular dysfunction, such as impaired energy metabolism, altered cell signaling and cell cycle control, impaired cell transport processes and dysfunctional biological activities, immune activation, and inflammation (Bozza et al. 2013). RONS can be involved in several disease processes as causative agents or result as an effect of the pathogenesis, including SAE.
Mason et al. developed an antibody that recognizes macromolecular DMPO (5,5-dimethyl-pyrroline-N-oxide) spin adducts, regardless of the oxidative/reductive state of the trapped radical adducts, and called the methodology immuno-spin trapping (IST) (Mason 2004; Ramirez and Mason 2005; Gomez-Mejiba et al. 2009; Khoo et al. 2015; Mason 2016; Gomez-Mejiba et al. 2014). DMPO is a spin trapping compound that has been used for several decades to trap and stabilize free radical species. Our group extended IST technique to an in vivo approach that involves the use IST in conjunction with molecular magnetic resonance imaging (mMRI) (Towner et al. 2012, 2013a, b, c, 2015; Coutinho de Souza et al. 2015; Towner and Smith 2017). The spin-trapping agent, DMPO, is used initially to trap free radicals in an oxidative stress-related disease model, and administration of a mMRI probe, called an anti-DMPO probe, that combines an antibody against DMPO-radical adducts and a MRI contrast agent, is used to target the DMPO-trapped free radicals that can be detected by mMRI. The anti-DMPO probe includes an albumin-Gd-DTPA-biotin construct, where the anti-DMPO antibody is covalently linked to the cysteine residues of albumin, forming an anti-DMPO-adduct antibody-albumin-Gd-DTPA-biotin entity. The Gd-DTPA moiety acts as the MRI signaling component, which will increase MRI SI in a T1-weighted morphological MR imaging sequence, and decrease T1 relaxation in a T1 map image. Both of these parameters, MRI SI or T1 relaxation, can be used to assess the presence of the anti-DMPO probe in vivo. In addition, the biotin moiety can be used for ex vivo validation of the presence of the anti-DMPO probe in tissues, by using streptavidin-fluorescence (e.g., Cy3) or streptavidin-HRP (horse radish peroxidase) to tag the biotin in the anti-DMPO probe.
We previously used the combined IST and mMRI free radical-targeted approach in a mouse model for SAE (Towner et al. 2013a). Mice with septic encephalopathy (induced by CLP) were found to have higher levels of trapped DMPO-radical adducts compared to sham animals (abdominal incision without CLP and sutured) (Towner et al. 2013a). This was the first reported in vivo detection of elevated in situ free radicals in a mouse model for SAE. However, long-term imaging findings associated with experimental sepsis were not addressed. In the current study, we used a combination of MR techniques, including CE-MRI, pMRI, and 1H–MRS and to assess long-term neuroinflammation-associated alterations in BBB permeability, microvascularity, and neurological metabolism, respectively, in a rat lipopolysaccharide (LPS)-induced SAE model over 3 months. Free radical-targeted mMRI was also used to assess brain free radical levels at 24 h and 1 week post-LPS injection.
Materials and methods
Ethics statement
Animal experiments were performed with the approval and strict adherence to the policies of the Oklahoma Medical Research Foundation Institutional Animal Care and Use Committee, which specifically approved this study, with adherence to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All efforts were made to minimize suffering.
LPS exposure
Rats (Sprague-Dawley; 8–10 weeks old; male; n = 30) were exposed to LPS (10 mg/kg in 100 μL saline; i.p.). Controls were administered saline (same volume and route of administration as for LPS).
MRI methods—contrast-enhanced MRI, perfusion imaging, and MR spectroscopy
MRI experiments were done on a Bruker Biospec 7.0 Tesla/30 cm horizontal-bore imaging system. Multiple brain 1H–MR image slices were taken using a RARE multi-slice (repetition time (TR) 1.3 s, echo time (TE) 9 ms, 256 × 256 matrix, 4 steps per acquisition, 4 × 4 cm2 field-of-view, 1.0-mm slice thickness) imaging sequence.
For contrast-enhanced MRI (CE-MRI), multi-slice spin echo T1-weighted images (TR = 1000.0 ms, TE = 14 ms, FOV = 4 × 4 cm2, averages = 2, slices = 16, matrix size = 256 × 256) were also performed and acquired before and 10, 20, and 30 min after intravenous contrast agent injection (Gd-DTPA, Magnevist, Bayer Inc., Wayne, NY, USA; 0.4 mmol/kg). Regional assessments were made in the following regions: cortex, perirhinal cortex (pr-cortex), hippocampus, and thalamus regarding MRI signal intensity measurements.
1H–MRS was acquired using a PRESS (Point REsolved SpectroScopy) sequence with a TE of 24.0 ms, a TR of 2500.0 ms, 512 averages, and a spectral width of 4006 Hz. A non-suppressed MR spectrum was acquired beforehand by applying eddy-current correction to maximize signal intensity and decrease the peak linewidths. Water was suppressed with a VAPOR (variable power radio frequency pulses and optimized relaxation delays) suppression scheme. In all cases, the peak width (full width at half maximum) of the water peak was less than 30 Hz following localized shimming, which was conducted by using first and second order adjustments with Fastmap. A cubic voxel of 3.0 × 3.0 × 3.0 mm3 was positioned in the rat brain. To analyze the MRS data, an in-house Mathematica program was used (version 6.0, Wolfram Research, Champaign, IL, USA). The spectra were scaled in ppm by calibrating against the water peak (4.78 ppm). The major brain metabolic peaks were identified as N-acetylaspartate (NAA) at 2.02 ppm, choline (Cho) at 3.22 ppm, creatine (Cr) at 3.02 ppm, and myo-inositol at 3.53 ppm. The peak area measurements of the metabolites were used to calculate the following ratios: NAA to Cho (NAA/Cho), Cr to Cho (Cr/Cho), and Myo-Ins to Cho (Myo-Ins/Cho).
Arterial spin-labeling (ASL) perfusion maps were obtained on a single axial slice of the brain located on the point of the rostro–caudal axis where the hippocampus had the largest cross-section. The imaging geometry was a 4 × 4 cm2 field-of-view (FOV) of 2 mm in thickness, with a single shot echo-planar encoding over a 64 × 64 matrix. An echo time (TE) of 13.5 ms, a repetition time (TR) of 18 s, and an inversion time (TIR) of 26.0 ms were used, and images were not submitted to time averaging. To obtain perfusion contrast, the flow alternating inversion recovery scheme was used. Briefly, inversion recovery images were acquired using a slice-selective inversion of the same geometry as the imaging slice or a nonselective inversion slice concentric with the imaging slice with a slice package margin of 5.0 mm. For each type of inversion, 22 images were acquired with inversion times evenly spaced from 26.0 to 8426.0 ms (with an increment of 400 ms between each TIR). Relative cerebral blood flow (rCBF) values were obtained by drawing circular ROIs (left and right regions) in the cortex, perirhinal cortex, hippocampus, and thalamus regions of the brain. Negative ASL rCBF values were assumed to be zero.
For free radical-targeted MRI, rat brains were imaged at 0 (pre-contrast) and up to 120 min post-contrast agent injection. Rats were previously treated with DMPO (5,5-dimethyl-1-pyrroline-N-oxide) (100 mg diluted in 200 μl saline; i.p.; 3× daily, every 6 h for 3 days) to trap free radicals, and subsequently injected intravenously with the anti-DMPO-albumin-Gd-DTPA-biotin contrast agent (200 μg anti-DMPO antibody/rat and 100 mg biotin-albumin-Gd-DTPA/rat) for in vivo free radical-targeted molecular MRI. T1-weighted images were obtained using a variable TR (repetition time) spin-echo sequence (TR, 200–1600 ms; TE, 15 ms; NA, 2). Pixel-by-pixel relaxation maps were reconstructed from a series of T1-weighted images using a nonlinear two-parameter fitting procedure. The T1 values of specified regions-of-interest (ROI) were computed from all the pixels in designated ROIs.
Streptavidin-HRP staining for anti-DMPO probe
Necropsies were performed on all rats. Brain tissues were fixed in 10% phosphate buffered formalin, embedded in paraffin, serially sectioned at 5 μm, and were stained with hematoxylin eosin (H&E) for histological examination. Streptavidin-HRP was used to target the biotin moiety of the anti-DMPO probe bound to macromolecular DMPO-trapped free radicals in excised rat brain tissues from the hippocampus region. Nova Red was used as the chromogen for HRP staining.
Statistical analyses
Statistical analyses were performed using Graph Pad Prism 6 (GraphPad Prism 6 Software, San Diego, CA, USA). All p values < 0.05 were considered statistically significant. MRI signal intensities, rCBF values, and metabolite peak ratios [(NAA/Cho), (Cr/Cho), and (Myo-Ins/Cho)] were reported as means ± standard deviations. For statistical analysis, Student’s t tests (independent samples, two-tailed t test) were used to assess the differences between means of the LPS-exposed and saline-control rat brains.
Results
LPS-treated rats have increased short-term and long-term blood-brain-barrier permeability alterations
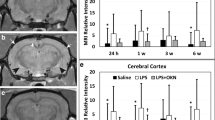
CE-MRI, which detects BBB permeability alterations, indicated a significant increase in MRI SI due to the presence of Gd-DTPA in LPS-exposed rat brains at 24 h post-injection in the cerebral cortex (p < 0.01), pr-cortex (p < 0.05), hippocampus (p < 0.01), and thalamus (p < 0.01), compared to saline-treated controls (Fig. 1a–d). Panel “iii” in Fig. 1 is a representative T1-weighted MR image of an LPS-exposed rat brain at 24 h post-LPS, compared to a saline-treated rat brain at the same time-point (Fig. 1(i)). Significantly increased MRI SI was also observed in the LPS rat brains in the cerebral cortex (p < 0.05), pr-cortex (p < 0.001), and hippocampus (p < 0.05) 1 week after LPS injection, compared to controls. The only long-term change to the BBB in LPS-exposed rats was detected in the cerebral cortex at 6 weeks following LPS injection (p < 0.01), compared to controls. Representative LPS-exposed (Fig. 1(iv)) and saline-treated (Fig. 1(i)) rat brain MR images at 6 weeks post-LPS are also shown. Cortex (1), hippocampus (2), thalamus (3), and pr-cortex (4) are numbered respectively.

LPS-treated rat brains have increased BBB permeability at early and late stages of LPS exposure. CE-MRI relative signal intensities for LPS- and saline-administered rat brains in the a cerebral cortex, b hippocampus, c thalamus, and d perirhinal cortex (pr-cortex) brain regions. Representative T1-weighted MR images of rat brains administered either saline (i, ii) or LPS (iii, iv) at either 24 h or 6 weeks post-LPS or saline. Regional assignments: (1) cerebral cortex, (2) hippocampus, (3) thalamus, and (4) pr-cortex. *p < 0.05, **p < 0.01, and ***p < 0.001
LPS-treated rats have both short-term and long-term altered brain tissue perfusion rates
pMRI indicated that for the cortex and thalamus regions, LPS-exposed rat brains had increased rCBF (p < 0.05 in the cortex, and p < 0.01 in the thalamus) at 24 h post-LPS, compared to controls; however, in the cortex and hippocampus, long-term significant decreases 6 weeks post-LPS in rCBF were detected for LPS-exposed rat brains (p < 0.001 for the cortex, and p < 0.01 for the hippocampus), compared to controls (Fig. 2). In the cortex, there were also significant decreases in rCBF at 3 and 12 weeks (p < 0.05 for both), compared to controls. Conversely, in the thalamus at 6 weeks post-LPS, there was a significant increase in rCBF in the LPS brains compared to controls. There were no significant changes in the perirhinal cortex regarding rCBF in LPS vs. controls (Fig. 2). Figure 2(iv) shows a representation perfusion map in a LPS-exposed rat brain 6 weeks post-LPS, compared to a saline-treated rat brain at the same time-point (Fig. 2(iii)). Morphological images for the perfusion maps shown in Fig. 2 are shown in panels “ii” for LPS and “i” for saline rat brains.

LPS-treated rat brains have altered rCBF at early and late stages of LPS exposure. rCBF values for LPS- and saline-administered rat brains in the a cerebral cortex, b hippocampus, c thalamus, and d perirhinal cortex brain regions. Representative T1-weighted MR images of rat brains administered either saline (i) or LPS (ii) at 6 weeks post-LPS or saline. Representative perfusion maps of rat brains administered either saline (iii) or LPS (iv) at 6 weeks post-LPS or saline. Regional assignments: (1) cerebral cortex, (2) hippocampus, (3) thalamus, and pr-cortex. *p < 0.05, **p < 0.01, and ***p < 0.001
LPS-treated rats have long-term altered brain metabolism
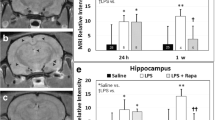
The MRS data indicated that LPS-exposed rat brains had significantly decreased NAA/Cho metabolite ratios at 1 (p < 0.05), 3 (p < 0.01), 6 (p < 0.05), and 12 (p < 0.001) weeks post-LPS injection; Cr/Cho ratios at 1, 3, and 12 weeks (p < 0.05 for all) post-LPS; and Myo-Ins/Cho ratios at 1, 3, and 6 weeks (p < 0.05 for all), compared to controls (Fig. 3a–c). Representative images depicting the selected regions for MR spectra in saline- and LPS-treated rat brains are shown in Fig. 3d(i, ii), respectively. Representative localized spectra for saline and LPS rat brains are shown in Fig. 3e.

LPS-treated rat brains have altered brain metabolism at early and late stages of LPS exposure. Metabolite/choline (Cho) ratios for a NAA/Cho, b Cr/Cho, and c Myo-Ins/Cho of LPS- and saline-administered rat brains. d Representative T1-weighted MR images of rat brains administered either saline (i) or LPS (ii) at 1 weeks post-LPS or saline. e Representative MR spectra from a saline- or LPS-administered rat brain. Peak assignments: myo-Ins at 4.06 ppm, tCho (total Cho) at 3.3 ppm, tCre (total Cr) at 3.0 ppm, and NAA at 2.0 ppm. *p < 0.05, **p < 0.01, and ***p < 0.001
LPS-treated rat brains have increased free radicals
The targeted free radical imaging indicated the detection of significantly increased trapped radical levels in LPS-exposed rat brains at both 24 h and 1 week post-LPS injection, compared to saline-treated controls (Fig. 4a–e). For the 24-h time-point, an increased uptake of the anti-DMPO probe (see Fig. 4f for illustration) in the hippocampal blood vessel endothelial cells (Fig. 4h) (compared to controls; Fig. 4g) was also observed, indicated by HRP staining due to the binding of streptavidin-HRP to the biotin moiety of the anti-DMPO probe. From the anti-DMPO probe streptavidin-HRP staining, it was also determined that there was an increase in blood vessel dilation at 24 h post-LPS (Fig. 4h).

LPS-treated rat brains have increased free radical levels at early stages of LPS exposure. Representative T1-weighted images overlaid with a post- minus a pre-contrast image following administration of the anti-DMPO probe that targets DMPO-trapped macromolecular radicals for a saline-, and LPS-administred rat brains at b 24 h and c 1 week post-LPS. MRI signal intensity percent (%) differences normalized to muscle graphs of LPS- and saline-administered rat brains given anti-DMPO probes at d 24 h or e 1 week post-LPS or saline. There was a significant increase in detection of DMPO-trapped radicals in the LPS-exposed rat brain at 24 h (p < 0.01) and 1 week (p < 0.001) post-LPS, compared to saline controls. f Illustration of the anti-DMPO probe, which consists of Gd-DTPA, anti-DMPO antibody bound to albumin, and biotin. Streptavidin-HRP bound to the biotin moiety of the anti-DMPO probe in a g saline- or h LPS-administered rat hippocampal brain tissue section. Note binding of the anti-DMPO probe to the endothelial cells, and more pronounced binding in the LPS brain, along with increased vessel dilation
Discussion
Our findings indicated that CE-MRI, which detects BBB permeability alterations, was able to show a significant increase in the uptake of Gd-DTPA in LPS-exposed rat brains at 24 h post-injection in the cerebral cortex (p < 0.01), pr-cortex (p < 0.05), hippocampus (p < 0.01), and thalamus (p < 0.01), compared to saline-treated controls (see Fig. 1). Significantly increased MRI SI was also observed in the LPS rat brains in the cerebral cortex (p < 0.05), pr-cortex (p < 0.001), and hippocampus (p < 0.05) 1 week after LPS injection, compared to controls. The only long-term change to the BBB in LPS-exposed rats was detected in the cerebral cortex at 6 weeks following LPS injection (p < 0.01), compared to controls.
It has been known for some time that LPS can disrupt the BBB (Wispelwey et al. 1988; Banks and Erickson 2010). It was recently found that Wip1 regulates BBB function and neuroinflammation induced by LPS via the sonic hedgehog signaling pathway (Zhen et al. 2017). In another study, dynamic contrast-enhanced (DCE) MRI was used to detect blood-brain barrier (BBB) breakdown 24 h following stereotaxic injection of LPS into the right striatum in rats, which was absent at 1 mon post-LPS injection (Ory et al. 2015). Another group used multiphoton imaging to detect increased BBB dysfunction in LPS-exposed mice (Zhou et al. 2014).
The cerebral cortex plays a key role in many brain functions, including memory, cognition, awareness, thought, language, attention, and consciousness (Shipp 2007). LPS exposure could possibly effect both short-term, as well as long-term cognition, which is currently being studied by our group members. The hippocampus is involved in both short-term and long-term memory (Squire 1992), which is also being investigated by our group in the context of LPS exposure. The thalamus is involved in the regulation of consciousness, alertness and sleep (Percheron 2003). The perirhinal cortex is thought to be associated primarily with memory (Moran and Dalrymple-Alford 2003) and is also currently being investigated by our group. Regarding altering BBB permeability, LPS exposure seems to affect all regions at early stages that we assessed, whereas only the cerebral cortex was observed to have more long-term effects. Increasing evidence indicates that the integrity of the BBB plays an important role regarding the onset and progression of neurodegeneration and cognitive impairment (Takechi et al. 2017).
Regarding brain tissue vascular perfusion, there were a few interesting occurrences that we observed (see Fig. 2). For the cortex and thalamus regions, LPS-exposed rat brains had increased rCBF (p < 0.05 in the cortex, and p < 0.01 in the thalamus) at 24 h post-LPS, compared to controls; however, in the cortex and hippocampus, long-term significant decreases 6 weeks post-LPS in rCBF were detected for LPS-exposed rat brains (p < 0.001 for the cortex, and p < 0.01 for the hippocampus), compared to controls. In the cortex, there were also significant decreases in rCBF at 3 and 12 weeks (p < 0.05 for both), compared to controls. Conversely, in the thalamus at 6 weeks post-LPS, there was a significant increase in rCBF in the LPS brains compared to controls. Another group assessing regional cerebral blood flow (CBF), measured by positron emission tomography (PET) using 18F–fluorodeoxyglucose (18F–FDG) in LPS-exposed rats, found CBF to decrease in the cortex (Semmler et al. 2008), which is similar to our findings. It is known that changes in cerebral blood flow, release of inflammatory molecules, and metabolic alterations, associated with SAE, have been previously found to contribute to neuronal dysfunction and cell death (Semmler et al. 2008). In a recent human study, cerebral perfusion alterations and cognitive decline were observed in critically ill sepsis survivors (Pierrakos et al. 2017). It is thought that alterations of CBF may represent a key component for the development of SAE (Taccone et al. 2013).
We also previously reported that vasogenic edematic fluid accumulates at the base of the brains of CLP-induced mice using MRI (Bozza et al. 2010). In a related study, evidence was found that there is a link between systemic inflammation and cerebral microhemorrhages (CMH), which are microscopic deposits of blood degradation products, and are pathological substrates of cerebral microbleeds (Sumbria et al. 2016). In the same study, with the use of Prussian blue staining, CMH was found to be significantly higher in LPS-treated C57BL/6 mice 7-days post-treatment, compared to saline-treated controls (Sumbria et al. 2016).
Our MRS studies indicated that LPS-exposed rat brains had significantly decreased NAA/Cho metabolite ratios at 1 (p < 0.05), 3 (p < 0.01), 6 (p < 0.05), and 12 (p < 0.001) weeks post-LPS injection; Cr/Cho ratios at 1, 3, and 12 weeks (p < 0.05 for all) post-LPS; and Myo-Ins/Cho ratios at 1, 3, and 6 weeks (p < 0.05 for all), compared to controls (see Fig. 3). It is interesting to note that in the LPS-induced SAE model, there were no significant decreases in metabolite ratios 24 h post-LPS; however, in a CLP-induced SAE mouse model we detected reduced NAA/Cho ratios at 24 h post-CLP (Bozza et al. 2010), possibly indicating that the CLP model is perhaps more severe than LPS injection. In another study, a NAA/Cr ratio was found to decrease in LPS-injected rat brains over a 24-h time-period (Wen et al. 2017). It was also found in the same study that the NAA/Cr ratio correlated quite well with apoptosis rates in the hippocampus (Wen et al. 2017).
Free radical imaging indicated detection of significantly increased trapped radical levels in LPS-exposed rat brains at both 24 h and 1 weeks post-LPS injection, compared to saline-treated controls (see Fig. 4). For the 24-h cohort, we also observed increased uptake of the anti-DMPO probe in the hippocampal blood vessel endothelial cells (compared to controls), indicated by HRP staining due to the binding of streptavidin-HRP to the biotin moiety of the anti-DMPO probe. From the anti-DMPO probe streptavidin-HRP staining, it was also observed that there was an increase in blood vessel dilation, which correlates with the increased rCBF at 24 h post-LPS. In our previous combined IST and mMRI study in a mouse SAE model (CLP-induced sepsis), we found that the distribution of the anti-DMPO probe was dispersed throughout the brain, and determined to be significantly higher in septic mice vs. sham animals in the hippocampus, striatum, occipital lobe and medial cortex regions of the brain, as measured by a percent (%) change or overall decrease in T1 relaxation (Towner et al. 2013a). Oxidized lipids (measured from Western blots for 4-hydroxynonenal (4-HNE)), and oxidized proteins (measured from Western blots for 3-nitroso-toluene (3-NT)) were also found to be significantly higher in septic mice (CLP) after 6 h, compared to sham controls (Towner et al. 2013a).
Others have also reported high levels of ROS were also detected in a mouse LPS-induced SAE model (Zhou et al. 2014; Clement et al. 2010). Increased ROS and nitric oxide (NO) were also detected in a feces-induced peritonitis sepsis mouse model (Handa et al. 2008). In another study using the CLP sepsis-induced mouse model, acute oxidative damage to the hippocampus was detected through increased 4-HNE expression, along with an increased Nox2 gene expression (Hernandes et al. 2014). Another report, using a LPS-induced SAE mouse model, showed high levels of ROS, malondialdehyde (MDA), tumor necrosis factor (TNF-α), and interleukin 1β (LI-1β) were all detected 8 h after LPS injection2. IL-6 was also found to be elevated in a LPS-induced SAE mouse model (Clement et al. 2010). Patients with encephalopathy were also found to have elevated serum and CSF (cerebral spinal fluid) levels of soluble intracellular adhesion molecule-1 (siCAM-1), a marker of endothelium-leukocyte interaction, nitric oxide (NO), lipid peroxide, and S100B, a marker of astrocyte activation and injury) compared to sepsis only (Hamed et al. 2009).
The pathogenesis of sepsis-induced brain dysfunction is associated with mitochondrial dysfunction (Bozza et al. 2013; Lyu et al. 2015; Wang et al. 2014). In a rat model of LPS-induced SAE, mitochondrial dysfunction was found to be associated with altered mitochondrial tyrosine kinase Src and protein phosphatase 1B (PTP1B) levels (Lyu et al. 2015). In the same study, pretreatment of mitochondrial proteins with active PTP1B resulted in overproduction of ROS and decreased mitochondrial membrane potential (Lyu et al. 2015). In a CLP-induced SAE mouse model, it was found that the mitochondrial function of the hippocampus was severely impaired, coupled with increased ROS, neuronal apoptosis and inflammation (Wu et al. 2015). In this same study, they investigated the effects of a mitochondria-targeted peptide SS-31 on mitochondrial function and cognitive deficits, and found that the peptide SS-1 protected mitochondrial integrity, reversed mitochondrial dysfunction, inhibited cytochrome c-related apoptosis, and diminished inflammation (Wu et al. 2015).
As there is mounting evidence that systemic inflammation coupled with aging may exacerbate cognitive decline (Sun et al. 2015; Yamanaka et al. 2017), an interesting future direction would be to assess long-term neuroinflammation and cognitive decline in aged rodents.
Conclusions
In a rat LPS endotoxemia model, MRI imaging demonstrates both acute and longer-term alterations in blood-brain barrier permeability, brain perfusion, metabolic alterations, and the generation of free radicals indicative of oxidative damage. MRI may provide a useful tool in the early identification of specific abnormalities associated with SAE and a mechanism to assess both the acute and sustained efficacy of mitigating therapies.
References
Banks WA, Erickson MA (2010) The blood-brain barrier and immune function and dysfunction. Neurobiol Dis 37(1):26–32. https://doi.org/10.1016/j.nbd.2009.07.031
Berg RM, Moller K, Bailey DM (2011) Neuro-oxidative-nitrosative stress in sepsis. J Cereb Blood Flow Metab 31(7):1532–1544. https://doi.org/10.1038/jcbfm.2011.48
Bettio LEB, Rajendran L, Gil-Mohapel J (2017) The effects of aging in the hippocampus and cognitive decline. Neurosci Biobehav Rev 79:66–86. https://doi.org/10.1016/j.neubiorev.2017.04.030
Bozza FA, Garteiser P, Oliveira MF, Doblas S, Cranford R, Saunders D, Jones I, Towner RA, Castro-Faria-Neto HC (2010) Sepsis-associated encephalopathy: a magnetic resonance imaging and spectroscopy study. J Cereb Blood Flow Metab 30(2):440–448. https://doi.org/10.1038/jcbfm.2009.215
Bozza FA, D’Avila JC, Ritter C, Sonneville R, Sharshar T, Dal-Pizzol F (2013) Bioenergetics, mitochondrial dysfunction, and oxidative stress in the pathophysiology of septic encephalopathy. Shock 39:10–16. https://doi.org/10.1097/SHK.0b013e31828fade1
Clement HW, Vasquez JF, Sommer O, Heiser P, Morawietz H, Hopt U, Schulz E, von Dobschutz E (2010) Lipopolysaccharide-induced radical formation in the striatum is abolished in Nox2 gp91phox-deficient mice. J Neural Transm (Vienna) 117(1):13–22. https://doi.org/10.1007/s00702-009-0327-5
Coutinho de Souza P, Smith N, Atolagbe O, Ziegler J, Nijoku C, Lerner M, Ehrenshaft M, Mason RP, Meek B, Plafker SM, Saunders D, Mamedova N, Towner RA (2015) OKN-007 decreases free radical levels in a preclinical F98 rat glioma model. Free Radic Biol Med 87:157–168. https://doi.org/10.1016/j.freeradbiomed.2015.06.026
Cunningham C, Hennessy E (2015) Co-morbidity and systemic inflammation as drivers of cognitive decline: new experimental models adopting a broader paradigm in dementia research. Alzheimers Res Ther 7(1):33. https://doi.org/10.1186/s13195-015-0117-2
Gao R, Ji MH, Gao DP, Yang RH, Zhang SG, Yang JJ, Shen JC (2017) Neuroinflammation-induced downregulation of hippocampal neuregulin 1-ErbB4 signaling in the parvalbumin interneurons might contribute to cognitive impairment in a mouse model of sepsis-associated encephalopathy. Inflammation 40(2):387–400. https://doi.org/10.1007/s10753-016-0484-2
Gomez-Mejiba SE, Zhai Z, Akram H, Deterding LJ, Hensley K, Smith N, Towner RA, Tomer KB, Mason RP, Ramirez DC (2009) Immuno-spin trapping of protein and DNA radicals: “tagging” free radicals to locate and understand the redox process. Free Radic Biol Med 46(7):853–865. https://doi.org/10.1016/j.freeradbiomed.2008.12.020
Gomez-Mejiba SE, Zhai Z, Della-Vedova MC, Muñoz MD, Chatterjee S, Towner RA, Hensley K, Floyd RA, Mason RP, Ramirez DC (2014) Immuno-spin trapping from biochemistry to medicine: advances, challenges, and pitfalls. Focus on protein-centered radicals. Biochim Biophys Acta 1840(2):722–729. https://doi.org/10.1016/j.bbagen.2013.04.039
Hamed SA, Hamed EA, Abdella MM (2009) Septic encephalopathy: relationship to serum and cerebrospinal fluid levels of adhesion molecules, lipid peroxides and S-100B protein. Neuropediatrics 40(2):66–72. https://doi.org/10.1055/s-0029-1231054
Handa O, Stephen J, Cepinskas G (2008) Role of endothelial nitric oxide synthase-derived nitric oxide in activation and dysfunction of cerebrovascular endothelial cells during early onsets of sepsis. Am J Physiol Heart Circ Physiol 295(4):H1712–H1719. https://doi.org/10.1152/ajpheart.00476.2008
Hernandes MS, D’Avila JC, Trevelin SC, Reis PA, Kinjo ER, Lopes LR, Castro-Faria-Neto HC, Cunha FQ, Britto LR, Bozza FA (2014) The role of Nox2-derived ROS in the development of cognitive impairment after sepsis. J Neuroinflammation 11(1):36. https://doi.org/10.1186/1742-2094-11-36
Jacob A, Brorson JR, Alexander JJ (2011) Septic encephalopathy: inflammation in man and mouse. Neurochem Int 58(4):472–476. https://doi.org/10.1016/j.neuint.2011.01.004
Jain NK, Patil CS, Kulkarni SK, Singh A (2002) Modulatory role of cyclooxygenase inhibitors in aging- and scopolamine or lipopolysaccharide-induced cognitive dysfunction in mice. Behav Brain Res 133(2):369–376. https://doi.org/10.1016/S0166-4328(02)00025-6
Khoo NK, Cantu-Medellin N, St Croix C, Kelley EE (2015) In vivo immuno-spin trapping: imaging the footprints of oxidative stress. Curr Protoc Cytom 74:12.42.1–12.4211
Lyu J, Zheng G, Chen Z, Wang B, Tao S, Xiang D, Xie M, Huang J, Liu C, Zeng Q (2015) Sepsis-induced brain mitochondrial dysfunction is associated with altered mitochondrial Src and PTP1B levels. Brain Res 1620:130–138. https://doi.org/10.1016/j.brainres.2015.04.062
Mason RP (2004) Using anti-5,5-dimethyl-1-pyrroline N-oxide (anti-DMPO) to detect protein radicals in time and space with immuno-spin trapping. Free Radic Biol Med 36(10):1214–1223. https://doi.org/10.1016/j.freeradbiomed.2004.02.077
Mason RP (2016) Imaging free radicals in organelles, cells, tissue, and in vivo with immuno-spin trapping. Redox Biol 8:422–429. https://doi.org/10.1016/j.redox.2016.04.003
Michels M, Danielslki LG, Vieira A, Florentino D, Dall’lgna D, Gallant L, Sonai B, Vuolo F, Mina F, Pescador B, Dominguini D, Barichello T, Quevedo J, Dal-Pizzol F, Petronilho F (2015) CD40-CD40 ligand pathway is a major component of acute neuroinflammation and contributes to long-term cognitive dysfunction after sepsis. Mol Med 21:219–226. https://doi.org/10.2119/molmed.2015.00070
Mikkelsen ME, Christie JD, Lanken PN, Biester RC, Thompson BT, Bellamy SL et al (2012) The adult respiratory distress syndrome cognitive outcomes study: long-term neuropsychological function in survivors of acute lung injury. Am J Respir Crit Care Med 185(12):1307–1315
Moran JP, Dalrymple-Alford JC (2003) Perirhinal cortex and anterior thalamic lesions: comparative effects on learning and memory. Behav Neurosci 117(6):1326–1341. https://doi.org/10.1037/0735-7044.117.6.1326
Murray C, Sanderson DJ, Barkus C, Deacon RM, Rawlins JN, Bannerman DM, Cunningham C (2012) Systemic inflammation induces acute working memory deficits in the primed brain: relevance for delirium. Neurobiol Aging 33(603–16):e3
Ning Q, Liu Z, Wang X, Zhang R, Zhang J, Yang M, Sun H, Han F, Zhao W, Zhang X (2017) Neurodegenerative changes and neuroapoptosis induced by systemic lipopolysaccharide administration are reversed by dexmedetomidine treatment in mice. Neurol Res 39(4):357–366. https://doi.org/10.1080/01616412.2017.1281197
Oedekoven CS, Jansen A, Keidel JL, Kircher T, Leube D (2015) The influence of age and mild cognitive impairment on associative memory performance and underlying brain networks. Brain Imaging Behav 9(4):776–789. https://doi.org/10.1007/s11682-014-9335-7
Ory D, Planas A, Dresselaers T, Gsell W, Postnov A, Celen S, Casteels C, Himmelreich U, Debyser Z, Van Laere K, Verbruggen A, Bormans G (2015) PET imaging of TSPOI in a rat model of local neuroinflammation induced by intracerebral injection of lipopolysaccharide. Nucl Med Biol 42(10):753–761. https://doi.org/10.1016/j.nucmedbio.2015.06.010
Percheron G (2003) Thalamus. In: Paxinos G, May J (eds) The human nervous system, 2nd edn. Elsevier, Amsterdam, pp 592–675
Pierrakos C, Attou R, Decorte L, Velissaris D, Cudia A, Gottignies P, Devriendt J, Tsolaki M, De Bels D (2017) Cerebral perfusion alterations and cognitive decline in critically ill sepsis survivors. Acta Clin Belg 72(1):39–44. https://doi.org/10.1080/17843286.2016.1191851
Ramirez DC, Mason RP (2005) Immuno-spin trapping: detection of protein-centered radicals. Curr Protoc Toxicol 17:17
Semmler A, Hermann S, Mormann F, Weberpals M, Paxian SA, Okulla T, Schafers M, Kummer MP, Klockgether T, Heneka MT (2008) Sepsis causes neuroinflammation and concomitant decrease of cerebral metabolism. J Neuroinflammation 5(1):38. https://doi.org/10.1186/1742-2094-5-38
Shipp S (2007) Structure and function of the cerebral cortex. Curr Biol 17(12):R443–R449. https://doi.org/10.1016/j.cub.2007.03.044
Squire LR (1992) Memory and the hippocampus: a synthesis from findings with rats, monkeys, and humans. Psychol Rev 99(2):195–231. https://doi.org/10.1037/0033-295X.99.2.195
Sumbria RK, Grigoryan MM, Vasilevko V, Krasieva TB, Scadeng M, Dvornikova AK, Paganini-Hill A, Kim R, Cribbs DH, Fisher MJ (2016) A murine model of inflammation-induced cerebral microbleeds. J Neuroinflammation 13(1):218. https://doi.org/10.1186/s12974-016-0693-5
Sun J, Zhang S, Zhang X, Zhang X, Dong H, Qian Y (2015) IL-17A is implicated in lipopolysaccharide-induced neuroinflammation and cognitive impairment in aged rats via microglial activation. J Neuroinflammation 12(1):165. https://doi.org/10.1186/s12974-015-0394-5
Taccone FS, Scolletta S, Franchi F, Donadello K, Oddo M (2013) Brain perfusion in sepsis. Curr Vasc Pharmacol 11(2):170–186
Takechi R, Lam V, Brook E, Giles C, Fimognari N, Mooranian A, Al-Salami H, Coulson SH, Nesbit M, Mamo JCL (2017) Blood-brain barrier dysfunction precedes cognitive decline and neurodegeneration in diabetic insulin resistant mouse model: an implication for causal link. Front Aging Neurosci 9:399. https://doi.org/10.3389/fnagi.2017.00399
Toth P, Tarantini S, Csiszar A, Ungvari Z (2017) Functional vascular contributions to gognitive impairement and dementia: mechanisms and consequences of cerebral autoregulatory dysfunction, endothelial impairment, and neurovascular uncoupling in aging. Am J Physiol Heart Circ Physiol 312(1):H1–H20. https://doi.org/10.1152/ajpheart.00581.2016
Towner R, Smith N (2017) In vivo and in situ detection of macromolecular free radicals using immune-spin trapping and molecular MRI. Antioxid Redox Signal. https://doi.org/10.1089/ars.2017.7390
Towner RA, Smith N, Saunders D, Henderson M, Downum K, Lupu F, Silasi-Mansat R, Ramirez DC, Gomez-Mejiba SE, Bonini MG, Ehrenshaft M, Mason RP (2012) In vivo imaging of immuno-spin trapped radicals with molecular MRI in a mouse diabetes model. Diabetes 61(10):2405–2413. https://doi.org/10.2337/db11-1540
Towner RA, Garteiser P, Bozza F, Smith N, Saunders D, d’Avila JCP, Magno F, Oliveira MF, Ehrenshaft M, Lupu F, Silasi-Mansat R, Ramirez DC, Gomez-Mejiba SE, Mason RP, Faria-Neto HCC (2013a) In vivo detection of free radicals in mouse septic encephalopathy using molecular MRI and immuno-spin-trapping. Free Radic Biol Med 65:828–837. https://doi.org/10.1016/j.freeradbiomed.2013.08.172
Towner RA, Smith N, Saunders D, De Souza PC, Henry L, Lupu F, Silasi-Mansat R, Ehrenshaft M, Mason RP, Gomez-Mejiba SE, Ramirez DC (2013b) Combined molecular MRI and immuno-spin-trapping for in vivo detection of free radicals in orthotopic mouse GL261 gliomas. Biochim Biophys Acta 1832(12):2153–2161. https://doi.org/10.1016/j.bbadis.2013.08.004
Towner RA, Smith N, Saunders D, Lupu F, Silasi-Mansat R, West M, Ramirez DC, Gomez-Mejiba SE, Bonini MG, Mason RP, Ehrenshaft M, Hensley K (2013c) In vivo detection of free radicals using molecular MRI and immuno-spin-trapping in a mouse model for amyotrophic lateral sclerosis. (ALS) Free Radic Biol Med 63:351–360. https://doi.org/10.1016/j.freeradbiomed.2013.05.026
Towner RA, Smith N, Saunders D, Carrizales J, Lupu F, Silasi-Mansat R, Ehrenshaft M, Mason RP (2015) In vivo targeted molecular magnetic resonance imaging of free radicals in diabetic cardiomyopathy within mice. Free Radic Res 49(9):1140–1146. https://doi.org/10.3109/10715762.2015.1050587
Wang Y, Chen Z, Zhang Y, Fang S, Zeng Q (2014) Mitochondrial biogenesis of astrocytes is increased under experimental septic conditions. Chin Med J 127(10):1837–1842
Wen M, Lian Z, Huang L, Zhu S, Hu B, Han Y, Deng Y, Zeng H (2017) Magnetic resonance spectroscopy for assessment of brain injury in the rat model of sepsis. Exp Ther Med 14(5):4118–4124. https://doi.org/10.3892/etm.2017.5034
Wispelwey B, Lesse AJ, Hansen EJ, Scheld WM (1988) Haemophilus influenza lipopolysaccharide-induced blood brain barrier permeability during experimental meningitis in the rat. J Clin Invest 82(4):1339–1346. https://doi.org/10.1172/JCI113736
Wu J, Zhang M, Hao S, Jia M, Ji M, Qui L, Sun X, Yang J, Li K (2015) Mitochondria-targeted peptide reverses mitochondrial dysfunction and cognitive deficits in sepsis-associated encephalopathy. Mol Neurobiol 52(1):783–791. https://doi.org/10.1007/s12035-014-8918-z
Yamanaka D, Kawano T, Nishigaki A, Aoyama B, Tateiwa H, Shigematsu-Locatelli M, Locatelli FM, Yokoyama M (2017) Preventive effects of dexmedetomidine on the development of cognitive dysfunction following systemic inflammation in aged rats. J Anesth 31(1):25–35. https://doi.org/10.1007/s00540-016-2264-4
Zhen H, Zhao L, Ling Z, Kuo L, Xue X, Feng J (2017) Wip1 regulates blood-brain barrier function and neuroinflammation induced by lipopolysaccharide via the sonic hedgehog signaling signaling pathway. Mol Immunol 93:31–37
Zhou T, Zhao L, Zhan R, He Q, Tong Y, Tian X, Wang H, Zhang T, Fu Y, Sun Y, Xu F, Guo X, Fan D, Han H, Chui D (2014) Blood-brain barrier dysfunction in mice induced by lipopolysaccharide is attenuated by dapsone. Biochem Biophys Res Commun 453(3):419–424. https://doi.org/10.1016/j.bbrc.2014.09.093
Ziaja M (2013) Septic encephalopathy. Curr Neurol Neurosci Rep 13(10):383. https://doi.org/10.1007/s11910-013-0383-y
Funding
Grant funding was provided by the National Institutes of Health (NIH) grant R01 NS092458.
Author information
Authors and Affiliations
Corresponding author
About this article

Cite this article
Towner, R.A., Saunders, D., Smith, N. et al. Assessing long-term neuroinflammatory responses to encephalopathy using MRI approaches in a rat endotoxemia model. GeroScience 40, 49–60 (2018). https://doi.org/10.1007/s11357-018-0009-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11357-018-0009-z