
Abstract
Chlorpyrifos (CPF) is a widely used organophosphate insecticide with several harmful effects. N-acetylcysteine (NAC) represents an ideal antixenobiotic; it can directly enter endogenous biochemical processes and is used as adjunctive treatment for psychiatric disorders. We aimed to evaluate the neuroprotective effect of NAC as an antioxidant drug against CPF-induced neurotoxicity in adult male albino rat brains. Twenty-eight male Wister rats were allocated into four groups (n = 7) and were administered the following for 28 days: group I (control group), physiological saline (0.9% NaCl); group II (CPF group), 10 mg/kg body weight (BW) CPF; group III (NAC group), 100 mg/kg BW NAC; and group VI (CPF+NAC group), NAC 1 h before CPF. CPF intoxication resulted in acetylcholinesterase inhibition, reduced glutathione content, and elevated levels of malondialdehyde and nitric oxide, which are oxidative stress biomarkers. CPF also depleted the activity of antioxidant enzymes, superoxide dismutase and catalase, and levels of inflammatory mediators, tumor necrosis factor-α, interleukin (IL)-6, and IL-1β. Levels of vascular endothelial growth factor, Bax, and the proapoptotic caspases-3 also increased, while brain-derived neurotrophic factor level decreased. Additionally, CPF significantly diminished Bcl-2 (an antiapoptotic protein) in rat brain cortical tissue. NAC treatment was found to protect brain tissue by reversing the CPF-induced neurotoxicity. Our results show the antioxidant, antiinflammatory, and antiapoptotic effects of NAC on CPF-induced neurotoxicity in rat brain tissue.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The wide and indiscriminate use of pesticides for agricultural, industrial, and public health purposes has caused serious environmental and health problems worldwide (Ross et al. 2013). Organophosphate pesticides (OPs) are a group of synthetic chemical compounds used in pesticides, and their toxicity to humans and other nontarget species has caused increasing concern (Maroni et al. 2000). OPs were found to have several harmful effects, including hepatic dysfunction, genotoxicity, teratogenicity, neurotoxicity, and neurobehavioral changes (Lassiter et al. 2010; Ncibi et al. 2008). Studies have indicated that OP toxicity signs are associated with the enhanced production of reactive oxygen species (ROS) (Ogut et al. 2011; Slotkin 2011).
Chlorpyrifos (O, O-diethyl-O-3,5, 6-trichloro-2-pyridyl phosphorothioate; CPF) is a lipophilic, broad-spectrum organophosphate insecticide, and is commercially used to control the foliar insects that affect crops and underground termites (Shou et al. 2019). The neuropsychological and psychiatric impairments in occupational workers exposed chronically to OPs have recently gained attention (Kaur et al. 2019). During occupational and residential exposure, pesticides are prevalently absorbed through the airways, and neurons are one of the targets of these compounds. In humans, several reports have suggested the involvement of the cholinergic pathway during acute or chronic exposure to OPs, which were accompanied by alterations in neuropsychological performance. This affected cognition and other correlated processes, such as processing speed, visuo-perceptual abilities, visual attention, memory impairment, and problem solving (Basaure et al. 2019). CPF poisoning inhibited acetylcholinesterase (AChE) activity, resulting in the accumulation of acetylcholine and neurochemical alternation, leading to death in severe cases (Al Omairi et al. 2019).
For several decades, NAC was available clinically as a mucolytic and antidote for acetaminophen poisoning. It has also been used for its clinically useful effects in treating numerous psychiatric disorders and neurological diseases (Finamor et al. 2014). NAC, a precursor of the amino acid l-cysteine, is a widely available and inexpensive drug (Mokhtari et al. 2017). In rats, oral NAC treatment has significantly ameliorated oxidative stress in phosphamidon, an organophosphate insecticide (Suke et al. 2008). Additionally, NAC has been shown to provide hepatorenal protection against Fipronil-induced toxicity in rats, via modulation of oxidative stress and apoptosis (Abdel-Daim et al. 2019). Moreover, NAC is now emerging as a treatment for vascular and nonvascular neurological disorders (Bhatti et al. 2017). Animal-based experiments have focused on the effects of NAC on several oxidative stress biomarkers and glutathione (GSH) levels (Pandya et al. 2014).
Studies investigating the effects of NAC on the disturbed antioxidant immune defense system in rat brains after chronic CPF treatment are scarce. Therefore, this study was conducted to evaluate the possible ameliorative effect of NAC as a precursor of intracellular GSH, and whether CPF induces neurotoxic effects. This was done by examining the biomarkers of oxidant/antioxidant status, in addition to levels of the preselected proinflammatory cytokines interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF)-α. Histopathological investigation and immunohistochemical analysis of B-cell lymphoma 2 (Bcl-2), Bcl-2-like protein 4 (Bax), and caspase-3 (Cas-3) as antiapoptotic/apoptotic markers were also carried out on the brain tissue of all treated rat groups. Furthermore, brain-derived neurotrophic factor (BDNF) as a marker for neurogenesis and vascular endothelial growth factors (VEGF) as a marker for vasculogenesis were estimated in the brains of rats that received long-term CPF treatment. This was done to monitor whether NAC potentiated neurogenesis and recovered damaged blood vessels, leading to amelioration and protection of rat brain tissue against CPF-induced neurotoxicity.
Materials and methods
CPF was obtained from Egyptian Company for Pesticides and Chemicals (EPIC), Cairo, Egypt. Prior to administration, CPF was diluted with distilled water. NAC was purchased from SEDICO Pharmaceutical Co. as effervescent instant sachets with a concentration of 600 mg, which dissolved in distilled water. A dosage of 100 mg/kg body weight (BW) was freshly prepared.
Experimental design
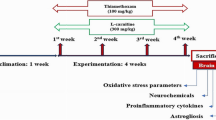
Twenty-eight adult male albino rats weighing 150–200 g (average 10 weeks old) were purchased from VACSERA (Cairo, Egypt). Animals were housed in polypropylene cages with controlled conditions (22 ± 2 °C and with a 12/12 h light/dark cycle). The study was approved, and all experimental procedures were performed by and according to the guidelines of the Committee of Research Ethics for Laboratory Animal Care, Department of Zoology and Entomology, Faculty of Science, Helwan University (Cairo, Egypt; approval no. HU2017/Z/05).
Animals were randomly divided into four experimental groups as follows: group I, serving as the control, received 10 ml/kg physiological saline (0.9% NaCl); group II, serving as the CPF-treated group, received 10 mg/kg of CPF; group III, serving as the NAC-administered group, received 100 mg/kg NAC solution; and group IV received a dose of NAC, followed by a dose of CPF after 1 h. Administration of either CPF and/or NAC was carried out daily for 28 consecutive days in each treated group. The animals of all groups were euthanized 24 h after the last administration, and blood was collected for further investigation. The brains were dissected, and half of each rat brain cortex in each group was used for biochemical investigations. The other half was prepared for histopathological and immunohistochemical investigation.
Biochemical analysis
Tissue homogenate preparation
Rat brain cortical tissue was homogenized in 50 mM Tris-HCl (pH 7.4) 10% (w/v), then centrifuged at 3000×g for 10 min at 4 °C. The obtained supernatants were used for all biochemical analyses. Total protein content in the supernatants was assessed according to the Lowry method [22].
Determination of acetylcholinesterase (AChE) activity
AChE activity was assayed as described by Ellman et al. (1961). Ellman’s colorimetric procedure was based on the reaction of thiocholine with 5,5-dithiobis-2-nitrobenzoic acid (DTNB, Ellman’s reagent), forming a yellow product (5-mercapto-2-nitrobenzoic acid and its dissociated forms) which was then measured at 412 nm.
Determination of oxidative stress markers
GSH content was determined using Ellman reagent, and the yellow chromagen formed was measured at 412 nm (Ellman 1959). Malondialdehyde (MDA), a marker for lipid peroxidation, was assayed using thiobarbituric acid, and the thiobarbituric acid-reactive substances formed were measured at 535 nm. These were then expressed in terms of MDA formed (Ohkawa et al. 1979). For estimating the amount of nitric oxide (NO), Griess’s reagent (sulfanilic acid and N-(1-naphthyl) ethylenediamine) was used, and the formed azo dye was measured at 540 nm (Green et al. 1982).
Determination of antioxidant enzyme activities
The activity of superoxide dismutase (SOD) was measured in terms of the capability of SOD to suppress the reduction of nitroblue tetrazolium (NBT) (Sun et al. 1988). Catalase (CAT) activity was assayed following the procedures described by Aebi (1984).
Determination of proinflammation markers
Pro-inflammatory cytokines (IL-1β, IL-6, and TNF-α) were measured using enzyme-linked immunosorbent assay (ELISA) kits purchased from ThermoFisher Scientific (IL-1,6; catalog number ERIL1B) and R&D Systems (TNF-α; catalog number RTA00), and levels were determined according to the manufacturers’ instructions. BDNF was estimated by ELISA using a kit purchased from RayBiotech Co., USA. VEGF was estimated by ELISA using a kit purchased from Invitrogen Co., CA, USA.
Histopathological examination
Tissue samples were fixed in 10% neutral-buffered formalin for 24 h at room temperature. They were dehydrated, embedded in Paraffin, sectioned (4–5 μm), and stained regularly with hematoxylin and eosin for light microscopy. A Nikon microscope (Eclipse E200-LED, Tokyo, Japan) was used for taking images, with an original magnification of × 400.
Immunohistochemical analysis
To investigate apoptosis-related proteins, the prepared brain samples were sectioned (4 μm thickness) and stained via immunohistochemistry with a rat polyclonal Bcl-2, Bax, and caspase-3 antibody. Images were taken with an original magnification of × 400 (Nikon Eclipse E200-LED, Tokyo, Japan).
Statistical analysis
Data are expressed as means ± standard deviation (SD). Data from various evaluations were examined by one-way analysis of variance and Duncan’s post hoc test, using a statistical package program (SPSS version 20.0).
Results
The present study aimed to explore the use of NAC as a strong antioxidant prodrug in protecting rat brains against CPF-induced neurotoxicity. As depicted in Fig.1, treatment of adult male rats with CPF for 28 consecutive days induced an overtly sharp and significant (p < 0.05) increase in AChE activity. This indicated the strong neurotoxic effect of CPF as an OP, as previously documented. Meanwhile, NAC treatment elevated AChE activity nonsignificantly in the NAC-treated group of rats. The administration of NAC to rats 1 h prior to CPF treatment for 28 consecutive days led to partial AChE activity elevation in the brain tissue of NAC+CPF-treated rats. However, there was a significant decrease in AChE (p < 0.05) compared to the control value.

The effect of N-acetylcysteine (NAC) on AChE activity in rat brain cortical tissue after oral administration of chlorpyrifos (CPF). Data are presented as mean ± SD (n = 7); a, p < 0.05 vs. control rats; b, p < 0.05 vs. NAC-treated rats using Duncan’s post hoc test
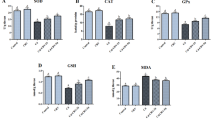
Oxidative stress in the brain tissue was determined in terms of MDA (end product of lipid peroxidation), NO, and enzymatic (SOD and CAT), and nonenzymatic (GSH) antioxidants. CPF induced oxidative stress, as evidenced by a significant (p < 0.05) elevation in MDA and NO levels, and a significant decrease (p < 0.05) in both GSH content and SOD and CAT activity (Figs. 2 and 3). These changes were observed in the brain tissue of CPF-treated rats, as compared to the control group values. Meanwhile, NAC-treated rat brains showed decreased MDA and NO levels, though this decrease was only significant (p < 0.05) for MDA. There was a nonsignificant increase in GSH content, and a significant (p < 0.05) decrease in SOD and CAT activity compared to the control group. On the other hand, the NAC+CPF-treated group indicated the strong antioxidant effect of NAC, as revealed in Fig. 2, with levels of MDA and NO levels changing significantly (p < 0.05). Meanwhile, a significant increase (p < 0.05) was evident in SOD activity, while CAT had a slight activity improvement when compared to the CPF-treated group. However, both enzymes significantly decreased (p < 0.05) when compared to the control values. The present results clearly demonstrate the effectiveness of NAC in protecting rat brains against oxidative stress resulting from CPF intoxication. This occurred by increasing GSH content and SOD activity and decreasing MDA and NO levels.

Effects of N-acetylcysteine (NAC) on MDA and NO levels in rat brain cortical tissue after oral administration of chlorpyrifos (CPF). Data are presented as mean ± SD (n = 7); a, p < 0.05 vs. control rats; b, p < 0.05 vs. NAC-treated rats using Duncan’s post hoc test

The effect of N-acetylcysteine (NAC) on enzymatic (SOD and CAT) and nonenzymatic (GSH) antioxidants in rat brain cortical tissue after oral administration of chlorpyrifos (CPF). Data are presented as mean ± SD (n = 7); a, p < 0.05 vs. control rats; b, p < 0.05 vs. NAC-treated rats using Duncan’s post hoc test
Investigation of the antiinflammatory effect of NAC on CPF-induced neurotoxicity was undertaken by measuring TNF-α, IL-1β, and IL-6 levels in rat brain tissue. The present results revealed that CPF treatment caused marked significant increases (p < 0.05) in TNF-α, IL-1β, and IL-6 levels (Fig. 4) in the brain tissue of the CPF-treated rats compared to control group values. However, NAC treatment did not lead to any changes in proinflammatory cytokine expression compared to control values. Data from the NAC+CPF-treated group indicated that NAC administration controlled the elevation of TNF-α, IL-1β, and IL-6 levels. There was a significant decrease (p < 0.05) in TNF-α, IL-1β, and IL-6 compared to the CPF-treated group, demonstrating the antiinflammatory activity of NAC against CPF neurotoxicity.

The effect of N-acetylcysteine (NAC) on proinflammatory cytokines (TNF-α, IL-1β, and IL-6) levels in rat brain cortical tissue after oral administration of chlorpyrifos (CPF). Data are presented as mean ± SD (n = 7); a, p < 0.05 vs. control rats; b, p < 0.05 vs. NAC-treated rats using Duncan’s post hoc test
To elucidate the effect of CPF-neurotoxicity on BDNF levels in rat brain tissue after 28 consecutive days of administration, an alteration in BDNF levels in the brain was detected in the CPF-treated group. This is illustrated in Fig. 5, with a significant decrease (p < 0.05) in BDNF compared to control values. NAC administration led to a significant increase (p < 0.05) in BDNF levels in the brain tissue of the NAC-treated group in comparison to the control values. Meanwhile, the NAC+CPF-treated group had significantly decreased (p < 0.05) brain BDNF levels, and showed an increase in the same parameter level being of nonsignificant change, if compared to CPF treated group.

The effect of N-acetylcysteine (NAC) on BDNF and VEGF levels in rat brain cortical tissue after oral administration of chlorpyrifos (CPF). Data are presented as mean ± SD (n = 7); a, p < 0.05 vs. control rats; b, p < 0.05 vs. CPF-treated rats using Duncan’s post hoc test
In addition to BDNF, VEGF levels were also determined in the brain tissue of all groups. The collected data is illustrated in Fig. 5, with a significant increase (p < 0.05) in VEGF levels observed in the brain tissue of CPF-treated rats compared to control group values. Meanwhile, VEGF elevated sharply and markedly, with a significant increase (p < 0.05) in NAC+CPF-treated rats compared to both control and CPF-treated group values. The NAC-treated group showed a slight nonsignificant increase in VEGF levels after 28 consecutive days of NAC administration.
The cortical tissue of the control and NAC-treated rats showed a normal characteristic structure (Fig. 6a, b, respectively). However, CPF-treated rats exhibited noticeable pyknotic and degenerated neurons, associated with the presence of many apoptotic neurons. A noteworthy finding was that inflammatory cell infiltration was also observed in the CPF-treated rats (Fig. 6b). NAC pretreatment improved the cortical deformations induced by CPF, though some neurons remained damaged (Fig. 6d).

Histopathological alternations in cortical tissue following treatment with N-acetylcysteine (NAC) and chlorpyrifos (CPF). a Photomicrograph of the cortical tissue of the control group showing normal cortical structure. b Photomicrograph of the cortical tissue of rats intoxicated with CPF showing degenerative neurons and leukocyte infiltration between neurons and apoptotic neurons. c Photomicrograph of the cortical tissue of rats treated with NAC alone showing a normal histological structure. d Photomicrograph of the cortical tissue of rats treated with NAC and CPF showing recovery of cortical tissue. However, some neurons show a degree of degeneration and less apoptotic neurons. Sections were stained with hematoxylin and eosin (× 400)
We examined Bcl-2, an antiapoptotic protein, and Bax, a proapoptotic protein, using immunohistochemistry to examine brain sections. The present results showed decreased intensity of immunoreactivity to Bcl-2. However, immunoreactivity to Bax was increased in CPF-treated rats (Figs. 7b and 8b, respectively). Conversely, preadministration with NAC greatly alleviated these conditions in the tested apoptotic biomarkers compared to CPF-treated rats. Thus, the results demonstrated that NAC has antiapoptotic action (Figs. 7d and 8d, respectively).

Cortical expression of Bcl-2 following the treatment with N-acetylcysteine (NAC) and/or chlorpyrifos (CPF) was evaluated using immunohistochemical staining. a represents the control group; b represents the CPF-treated group; c represents the NAC-treated group; and d represents the NAC+CPF-treated group (× 400)

Cortical expression of Bax following treatment with N-acetylcysteine (NAC) and/or chlorpyrifos (CPF) was evaluated using immunohistochemical staining. a represents the control group; b represents the CPF-treated group; c represents the NAC-treated group; and d represents the NAC+CPF-treated group (× 400)
Discussion
The widespread use of different pesticides in agriculture and for purposes of public health has led to severe effects on humans. One of these pesticides is the OP insecticide CPF, which is used for controlling different types of insects. It is also used on many types of crops and plants (Ross et al. 2013). Environmental, occupational, and food exposure have been associated with health risks, as multi-residues of pesticides has been found in nonorganic fruit, vegetables, and honey (Garcia-Garcia et al. 2016). Growing evidence has indicated the involvement of environmental chemicals in toxicity during brain development, leading to neurobehavioral changes such as learning disabilities, attention deficit hyperactivity disorder, cognitive impairment, spatial memory damage, autism, and neurodegenerative diseases (Abdel Moneim 2013; Landrigan et al. 2012).
The present study aimed to evaluate the role of NAC on CPF-induced neurotoxicity in the brain tissue of adult male rats. This was done by investigating oxidant/antioxidant status, inflammatory status, apoptotic/antiapoptotic markers, and factors responsible for neurogenesis. This was intended to minimize the oxidative stress of CPF, therefore minimizing brain damage. In this study, CPF decreased AChE activity and GSH content, and elevated the levels of oxidative stress markers MDA and NO in the brains of CPF-treated rats. However, SOD and CAT activity significantly decreased after CPF treatment compared with the control values. Previous studies showed that CPF treatment generates oxidative stress in rats and different cell models, as it easily diffuses through the cell membrane (Basha and Poojary 2011). CPF was documented to enhance cholinergic activity by AChE inhibition. It therefore has an anticholinesterase effect, leading to the inhibition of brain AChE at synapses, acetylcholine accumulation, and overactivation of the acetylcholine receptor at the neuromuscular junction and in the autonomic and central nervous system. In addition, previous studies have indicated that cognitive and emotional disorders are mediated by AChE inhibition. AChE is an enzyme that degrades acetylcholine in laboratory animals after CPF exposure (Cardona et al. 2011; Cardona et al. 2013).
Any sudden or chronic overconsumption of oxygen leads to ROS production, which may occur either in the mitochondria or inside the capillary system. It occurs as an oxidative burst induced by inflammatory cells. About 2–5% of oxygen in the mitochondrial electron transport system results in superoxide production, which is the most known of the free radicals. It is commonly produced during oxidative phosphorylation (Kerksick and Willoughby 2005). ROS are produced primarily by the mitochondria as a by-product of normal cell metabolism during the conversion of molecular oxygen (O2) to water (H2O). These include superoxide radical (O2__), hydrogen peroxide (H2O2), and hydroxyl radical (_OH). Peroxisomes produce H2O2 during fatty acid degradation. H2O2 is mostly degraded into water by catalase, but some molecules may also escape into the cell (Abdel Moneim 2016). When ROS production overwhelms antioxidant status, oxidative stress occurs, leading to lipid peroxidation and destruction of the macromolecules of cells and tissues (Nita and Grzybowski 2016).
In the present study, increased MDA and NO content in the brains of CPF-treated rats agree with previously documented results, demonstrating that this leads to apoptotic cell death in neuronal cells, thus causing neuronal injury (Almeer et al. 2018). It has been proposed that oxidative stress induction may also be involved in NO-induced apoptosis (Troy et al. 1996). The antioxidant defense system protects cells from attack by ROS. There are several antioxidant defense mechanisms; however, both oxidants and antioxidants have a profound effect on gene expression. Cells are equipped with several antioxidant agents, including enzymes such as SOD, CAT, and glutathione peroxidase (Yu et al. 2000) as well as endogenous thiols, or sulfhydryl containing compounds, such as GSH. GSH is the unique source of the thiol pool in the body, preventing damage to biological macromolecules caused by ROS. It plays many roles in the body, such as antioxidant defense, detoxification of electrophilic xenobiotics, regulating signal transduction antioxidant defense, neurotransmitter signaling, cysteine storage and transport, cell proliferation regulation, deoxyribonucleotide synthesis, immune response regulation, and leukotriene and prostaglandin metabolism. In addition, GSH has an important role in maintaining the redox state of the cell (Almeer et al. 2018), thereby exerting a profound protective effect on cells. Of the three amino acids (glutamate, glycine, and cysteine) in the GSH structure, cysteine has the lowest intracellular concentration (Aruoma et al. 1989).
In the present study, GSH levels were significantly decreased by CPF treatment and lipid peroxidation resulting from CPF neurotoxicity, and was associated with decreased cellular antioxidants, such as SOD and CAT (Astiz et al. 2012). Recently, CPF was found to modify the oxidative status in vitro by increasing ROS levels (Quintana et al. 2018), while at higher concentrations, it only decreased CAT and did not modify SOD activity. CPF decreased Krebs cycle enzyme activity levels (Basha and Poojary 2014) and severely depleted manganese levels, which is associated with Mn-SOD suppression and mitochondrial dysfunction (Samsel and Seneff 2015). In response to the cellular antioxidant systems of CPF, Lee et al. (2012) found that CPF treatment reduced the expression of CuZn-SOD and Mn-SOD.
The ability of a compound to cross the blood–brain barrier (BBB) is thought to be critical in targeting brain dysfunction for treatment. Despite NAC’s poor penetration into the CNS and GSH deficiency, earlier reports have supported the role of GSH in the effects exerted by NAC. As GSH levels in the brain subjected to oxidative stress were elevated after NAC treatment, this was considered to be the key to NAC action (das Neves Duarte et al. 2012). Cysteine was found to limit the rate of GSH synthesis during oxidative stress, as the liver of mammals tightly regulates free cysteine. Repeated doses of NAC were needed to affect its circulating free level and to be the most effective (Stipanuk et al. 2006). The uniqueness of NAC owes to the fact that this drug is an excellent source of sulfhydryl groups, and is thus an acetylated cysteine precursor of the amino acid l-cysteine. This sustains GSH synthesis by serving as an l-cysteine residue, and is converted in vivo into metabolites that stimulate glutathione production. It thereby maintains intracellular GSH levels, enhances detoxification, and acts directly as a free-radical scavenger. This is because it interacts with ROS such as OH_ and H2O2, thereby increasing cell protection against oxidative stress (Almeer et al. 2018). The present study indicated that NAC treatment of CPF-treated rats increased GSH levels and SOD activity. NAC effectively ameliorated GSH reduction in the brains of CPF-treated rats and increased the activity of SOD. However, it did not restore CAT activity to the control value. The present study documents the protective role of NAC in the brains of CPF-treated rats. This protective effect occurs through GSH production and establishing SOD activity. NAC administration during CPF intoxication showed its direct reaction with ROS, causing an increase in SOD activity and lowering the brain’s susceptibility to these oxidants (Farbiszewski et al. 2000).
CPF also inhibited mitochondrial oxidative phosphorylation and induced apoptosis in human neuroblastoma SH-SY5Y cells, or human neural precursor cells (Lee et al. 2014). Moreover, some endogenous compounds, including reactive oxygen species, lipid metabolites such as sphingosine and phosphatidic acid, and some endogenous cell death effectors such as Bax, can also cause lysosomal membrane permeabilization (LMP). This was reported to occur as a result of osmotic lysis or detergent activity of the compounds that accumulate in the lumen of lysosomes. LMP can either initiate or amplify different types of lysosomal cell death, including nonprogrammed (accidental) necrosis, lysosomal apoptosis, or cell death with apoptosis-like features. This can be initiated by the release of cathepsins and other hydrolases to the cytosol (Repnik et al. 2014).
In the present study, an increase in the levels of proinflammatory cytokines TNF-α, IL-1β, and IL-6 in the rat brains was recorded in the CPF-treated group. These increments in TNF-α, IL-1β, and IL-6 levels after CPF exposure were due to the activation of acute phase inflammatory responses, leading to release of these cytokines involved in the generation of ROS via mitochondrial respiratory chain reaction (Duramad et al. 2007). Due to the production of ROS, which damage lipids, proteins, and DNA, these mediators were also involved in various biological and cellular responses. These include tumor progression, transcription factor production, growth factor production, and activation of proapoptotic proteins (Abdel Moneim 2016). Moreover, astrocyte impairment was reported to be accompanied by increased TNF-α, IL-1β, and IL-6 secretion, which was associated with brain inflammation development (Wang et al. 2015).
ROS induction by pesticides occurs mainly through mitochondrial destabilization, accompanied by paralleled DNA damage. Many pathways, such as interaction with excitatory amino acid receptors, depletion of cellular NAD+, and activation of caspases, were involved in a cascade of events leading to NO-induced apoptosis (Abdel Moneim 2013). Bcl-2 is widely recognized as an antiapoptotic and antioxidant compound, and has an impact on the mitochondria. This is because binding of Bcl-2 and its antiapoptotic relatives to Bax blocks its oligomerization, thus conserving mitochondrial membrane integrity and cell survival. On the other hand, Bax pierces the outer mitochondrial membrane, mediating cell death by apoptosis by releasing apoptogenic proteins into the cytoplasm (Abdel Moneim 2016). Cas-3 is a frequently enhanced death protease, catalyzing the specific cleavage of many key cellular proteins (Moneim 2015). In the present study, CPF-induced apoptotic neuronal cell death occurred by elevating Bax and Cas-3-mRNA levels and diminishing Bcl-2 levels. However, pretreatment with NAC led to reduced Bax and Cas-3 and aggregated Bcl-2, indicating neuroprotection. Hence, NAC protected rats against neuronal injury by modulating apoptotic regulatory proteins and Cas-3 in CPF-induced neuronal apoptosis.
Earlier studies have indicated that NAC treatment is associated with decreased levels of IL-1β, TNF-α, IL-6, NF-κB, astrocyte injury marker, beta-amyloid precursor protein, and neurofilament light (a marker of axonal injury) (Chen et al. 2008). Due to the reaction of NAC with ROS and formaldehyde, their toxicity is diminished. NAC also acts as a donor of sulfhydryl groups, stabilizing the membrane lipid-protein structure, as previously reported by Farbiszewski et al. (2000), and NAC treatment thus decreases TNF-α, IL-1β, and IL-6. Moreover, NAC improved markers which modulate oxidative stress, such as apoptosis-related proteins Bcl-2 and Bax proteins, and the protein involved in the oxidative stress cascade (hemeoxygenase-1). NAC also increased the levels of membrane proteins involved in neurotransmission (Complexin I and II) (Yi et al. 2006).
NAC was found to reduce B cell lymphoma apoptosis by increasing concanavalin A-induced mitogenesis, blocking the lipopolysaccharide-induced apoptosis of endothelial cells. It also blocked LDL-induced superoxide production and the apoptosis of human umbilical vein endothelial cells (Sun et al. 2012). Through ROS inhibition, NAC inhibited in vitro apoptosis of Arabinoside neurotoxicity (Geller et al. 2001), supporting neuronal survival. According to Lee et al. (2012), NAC treatment attenuated mitochondrial inhibition and blocked apoptosis via the caspase-9 and caspase-3 pathways.
NAC was reported to be used as a treatment of vascular and nonvascular neurological disorders, also modulating glutamatergic, neurotrophic, and inflammatory pathways, thus healing brain dysfunctions and neuropathies (Bavarsad Shahripour et al. 2014). The results of the present study demonstrated a significant increase in VEGF levels in CPF-treated rat brains compared to the normal control group. This could be attributed to the toxicant-evoked hypoxia-like responses, followed by VEGF expression and activation, as documented earlier by Kim et al. (2012). VEGF is an angiogenic factor required for vasculature development and is a potent inducer of vascular permeability, a survival factor for newly formed blood vessels, and an inducer of pathologic angiogenesis (Yao et al. 2010). In the present study, it was demonstrated that NAC did not affect angiogenesis on its own. The results of the present study revealed that NAC administration to CPF-treated rats led to increased levels of VEGF in rat brain tissue compared with the control group or CPF-treated animals. Histological examination indicated greater levels of angiogenesis in the brains of both CPF and NAC+CPF-treated rats, which could be attributed to increased blood flow to the target inflamed cells for catching ROS. Recently, Zhang et al. (2018) suggested the antagonism of NAC on inflammation-induced pathological angiogenesis in a lipopolysaccharide-induced model of angiogenesis in chick chorioallantoic membranes. This study led to upregulation of the genes involved in tight-junction adhesion molecules, and also modulation of the subcellular distribution of junction and cell polarity proteins. This resulted in junction formation and epithelial polarization. The expression of these genes was raised following LPS exposure, and most of these genes’ expression dropped with the addition of NAC. Moreover, the authors also suggested that NAC, as an antioxidant, was able to restore pathological angiogenesis at least partially by the modulation of genes involved in oxidative stress.
The results of the present study demonstrated a significant decrease in BDNF levels in the brains of CPF group rats in comparison to the control group. This decrease in BDNF was suggested to be due to the injury of nerves and glial cells in the rat brain because of increased ROS production (Blanco et al. 2011). BDNF is a dimeric protein belonging to the neurotrophin family of survival-promoting molecules. It is found throughout the brain, with particular abundance in the hippocampus and cerebral cortex. BDNF was suggested to play an important role in neuronal survival, regeneration following injury, regulation of transmitter systems, and in the attenuation of neural immune responses. BDNF modulates synaptic plasticity and neurotransmitters, intracellular signal-transduction pathways, and regulates axonal and dendritic branching. It regulates re-modeling synaptogenesis in axon terminals, synaptic transmission, and is involved in the functional maturation of excitatory and inhibitory synapses. It was evidenced that BDNF may play important roles in mood disorder pathogenesis and in the action of therapeutic agents concerning mood and antidepressants. BDNF may also be involved in the pathophysiology of anxiety disorders (Liu et al. 2019).
NAC was found to improve cognitive deficits and neurogenesis. This is consistent with the hypothesis of NAC’s facilitated selective delivery to affected sites due to vascular effects, as reported in traumatic brain injury (Karalija et al. 2012). Roth et al. (2014) reported glutathione entrance into the brain from the periphery, exerting neuroprotective activity. The observed alterations in BDNF expression in different brain areas at different time points may represent normal stress responses aimed at restoring homeostasis. The present study attributes BDNF increase to the ability of NAC as free radical scavenger and antioxidant, used in inhibiting the progression of neurodegenerative disorders (Karalija et al. 2012). This assists with eliminating free radicals, which are the first cause for the ROS liberation causing apoptosis and rat brain injury due to CPF treatment. NAC showed an ability to play a neuroprotective role against neural damage and as an antioxidant to reduce brain tissue inflammation, contributing positively to the high increase in brain levels of BDNF (Hicdonmez et al. 2006).
Conclusions
The long-term treatment of rats with CPF induced oxidative stress and decreased GSH content in the brain. These changes were ameliorated by NAC, as evidenced by the observed decreases in MDA and NO levels, improved SOD activity and GSH content, the modulation of the inflammatory response, and the resulting antiapoptotic effects. The present findings demonstrate the neuroprotective and neurogenesis-promoting effects of NAC in the brain tissue of rats exposed to CPF-induced neurotoxicity.
References
Abdel Moneim AE (2013) The neuroprotective effects of purslane (Portulaca oleracea) on rotenone-induced biochemical changes and apoptosis in brain of rat. CNS Neurol Disord Drug Targets 12(6):830–841
Abdel Moneim AE (2016) Indigofera oblongifolia prevents lead acetate-induced hepatotoxicity, oxidative stress, fibrosis and apoptosis in rats. PLoS One 11:e0158965
Abdel-Daim MM, Dessouki AA, Abdel-Rahman HG, Eltaysh R, Alkahtani S (2019) Hepatorenal protective effects of taurine and N-acetylcysteine against fipronil-induced injuries: the antioxidant status and apoptotic markers expression in rats. Sci Total Environ 650:2063–2073
Aebi H (1984) Catalase in vitro. Methods Enzymol 105:121–126
Al Omairi NE, Al-Brakati AY, Kassab RB, Lokman MS, Elmahallawy EK, Amin HK, Abdel Moneim AE (2019) Soursop fruit extract mitigates scopolamine-induced amnesia and oxidative stress via activating cholinergic and Nrf2/HO-1 pathways. Metab Brain Dis. https://doi.org/10.1007/s11011-019-00407-2
Almeer RS, Kassab RB, AlBasher GI, Alarifi S, Alkahtani S, Ali D, Abdel Moneim AE (2018) Royal jelly mitigates cadmium-induced neuronal damage in mouse cortex. Mol Biol Rep 46(1):119–131
Aruoma OI, Laughton MJ, Halliwell B (1989) Carnosine, homocarnosine and anserine: could they act as antioxidants in vivo? Biochem J 264:863–869
Astiz M, de Alaniz MJ, Marra CA (2012) The oxidative damage and inflammation caused by pesticides are reverted by lipoic acid in rat brain. Neurochem Int 61:1231–1241
Basaure P, Guardia-Escote L, Cabre M, Peris-Sampedro F, Sanchez-Santed F, Domingo JL, Colomina MT (2019) Learning, memory and the expression of cholinergic components in mice are modulated by the pesticide chlorpyrifos depending upon age at exposure and apolipoprotein E (APOE) genotype. Arch Toxicol 93(3):693–707
Basha PM, Poojary A (2011) Chlorpyrifos induced region specific vulnerability in rat CNS and modulation by age and cold stress: an interactive study. Neurochem Res 36:241–249
Basha PM, Poojary A (2014) Mitochondrial dysfunction in aging rat brain regions upon chlorpyrifos toxicity and cold stress: an interactive study. Cell Mol Neurobiol 34:737–756
Bavarsad Shahripour R, Harrigan MR, Alexandrov AV (2014) N-acetylcysteine (NAC) in neurological disorders: mechanisms of action and therapeutic opportunities. Brain Behav 4:108–122
Bhatti J, Nascimento B, Akhtar U, Rhind SG, Tien H, Nathens A, da Luz LT (2017) Systematic review of human and animal studies examining the efficacy and safety of N-acetylcysteine (NAC) and N-acetylcysteine amide (NACA) in traumatic brain injury: impact on neurofunctional outcome and biomarkers of oxidative stress and inflammation. Front Neurol 8:744
Blanco J, Mulero M, Lopez M, Domingo JL, Sanchez DJ (2011) BDE-99 deregulates BDNF, Bcl-2 and the mRNA expression of thyroid receptor isoforms in rat cerebellar granular neurons. Toxicology 290:305–311
Cardona D, Lopez-Crespo G, Sanchez-Amate MC, Flores P, Sanchez-Santed F (2011) Impulsivity as long-term sequelae after chlorpyrifos intoxication: time course and individual differences. Neurotox Res 19:128–137
Cardona D, Lopez-Granero C, Canadas F, Llorens J, Flores P, Pancetti F, Sanchez-Santed F (2013) Dose-dependent regional brain acetylcholinesterase and acylpeptide hydrolase inhibition without cell death after chlorpyrifos administration. J Toxicol Sci 38:193–203
Chen G, Shi J, Hu Z, Hang C (2008) Inhibitory effect on cerebral inflammatory response following traumatic brain injury in rats: a potential neuroprotective mechanism of N-acetylcysteine. Mediat Inflamm 716458
das Neves Duarte JM, Kulak A, Gholam-Razaee MM, Cuenod M, Gruetter R, Do KQ (2012) N-acetylcysteine normalizes neurochemical changes in the glutathione-deficient schizophrenia mouse model during development. Biol Psychiatry 71:1006–1014
Duramad P, Tager IB, Holland NT (2007) Cytokines and other immunological biomarkers in children's environmental health studies. Toxicol Lett 172:48–59
Ellman GL (1959) Tissue sulfhydryl groups. Arch Biochem Biophys 82:70–77
Ellman GL, Courtney KD, Andres V Jr, Feather-Stone RM (1961) A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 7:88–95
Farbiszewski R, Witek A, Skrzydlewska E (2000) N-acetylcysteine or trolox derivative mitigate the toxic effects of methanol on the antioxidant system of rat brain. Toxicology 156:47–55
Finamor IA, Ourique GM, Pes TS, Saccol EM, Bressan CA, Scheid T, Baldisserotto B, Llesuy SF, Partata WA, Pavanato MA (2014) The protective effect of N-acetylcysteine on oxidative stress in the brain caused by the long-term intake of aspartame by rats. Neurochem Res 39:1681–1690
Garcia-Garcia CR, Parron T, Requena M, Alarcon R, Tsatsakis AM, Hernandez AF (2016) Occupational pesticide exposure and adverse health effects at the clinical, hematological and biochemical level. Life Sci 145:274–283
Geller HM, Cheng KY, Goldsmith NK, Romero AA, Zhang AL, Morris EJ, Grandison L (2001) Oxidative stress mediates neuronal DNA damage and apoptosis in response to cytosine arabinoside. J Neurochem 78:265–275
Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR (1982) Analysis of nitrate, nitrite, and [15 N]nitrate in biological fluids. Anal Biochem 126:131–138
Hicdonmez T, Kanter M, Tiryaki M, Parsak T, Cobanoglu S (2006) Neuroprotective effects of N-acetylcysteine on experimental closed head trauma in rats. Neurochem Res 31:473–481
Karalija A, Novikova LN, Kingham PJ, Wiberg M, Novikov LN (2012) Neuroprotective effects of N-acetyl-cysteine and acetyl-L-carnitine after spinal cord injury in adult rats. PLoS One 7:e41086
Kaur S, Singla N, Dhawan DK (2019) Neuro-protective potential of quercetin during chlorpyrifos induced neurotoxicity in rats. Drug Chem Toxicol:1–11
Kerksick C, Willoughby D (2005) The antioxidant role of glutathione and N-acetyl-cysteine supplements and exercise-induced oxidative stress. J Int Soc Sports Nutr 2:38–44
Kim J, Lim W, Ko Y, Kwon H, Kim S, Kim O, Park G, Choi H (2012) The effects of cadmium on VEGF-mediated angiogenesis in HUVECs. J Appl Toxicol 32:342–349
Landrigan PJ, Lambertini L, Birnbaum LS (2012) A research strategy to discover the environmental causes of autism and neurodevelopmental disabilities. Environ Health Perspect 120:a258–a260
Lassiter TL, Ryde IT, Levin ED, Seidler FJ, Slotkin TA (2010) Neonatal exposure to parathion alters lipid metabolism in adulthood: interactions with dietary fat intake and implications for neurodevelopmental deficits. Brain Res Bull 81:85–91
Lee JE, Park JH, Shin IC, Koh HC (2012) Reactive oxygen species regulated mitochondria-mediated apoptosis in PC12 cells exposed to chlorpyrifos. Toxicol Appl Pharmacol 263:148–162
Lee JE, Park JH, Jang SJ, Koh HC (2014) Rosiglitazone inhibits chlorpyrifos-induced apoptosis via modulation of the oxidative stress and inflammatory response in SH-SY5Y cells. Toxicol Appl Pharmacol 278:159–171
Liu J, Zhu HX, Fu WL, Xu XW, Yang JZ, Dai D, Li Y (2019) Downregulated hippocampal expression of brain derived neurotrophic factor and tyrosine kinase B in a rat model of comorbid epilepsy and depression. Neurol Res:1–9
Maroni M, Colosio C, Ferioli A, Fait A (2000) Biological monitoring of pesticide exposure: a review. Introduction Toxicology 143:1–118
Mokhtari V, Afsharian P, Shahhoseini M, Kalantar SM, Moini A (2017) A review on various uses of N-acetyl cysteine. Cell J 19:11–17
Moneim AE (2015) Oxidant/antioxidant imbalance and the risk of Alzheimer's disease. Curr Alzheimer Res 12:335–349
Ncibi S, Ben Othman M, Akacha A, Krifi MN, Zourgui L (2008) Opuntia ficus indica extract protects against chlorpyrifos-induced damage on mice liver. Food Chem Toxicol 46:797–802
Nita M, Grzybowski A (2016) The role of the reactive oxygen species and oxidative stress in the pathomechanism of the age-related ocular diseases and other pathologies of the anterior and posterior eye segments in adults. Oxidative Med Cell Longev 3164734
Ogut S, Gultekin F, Kisioglu AN, Kucukoner E (2011) Oxidative stress in the blood of farm workers following intensive pesticide exposure. Toxicol Ind Health 27:820–825
Ohkawa H, Ohishi N, Yagi K (1979) Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem 95:351–358
Pandya JD, Readnower RD, Patel SP, Yonutas HM, Pauly JR, Goldstein GA, Rabchevsky AG, Sullivan PG (2014) N-acetylcysteine amide confers neuroprotection, improves bioenergetics and behavioral outcome following TBI. Exp Neurol 257:106–113
Quintana MM, Rivero Osimani V, Magnarelli G, Rovedatti MG, Guinazu N (2018) The insecticides chlorpyrifos and acetamiprid induce redox imbalance in umbilical cord blood erythrocytes in vitro. Pestic Biochem Physiol 148:87–92
Repnik U, Hafner Cesen M, Turk B, 2014 Lysosomal membrane permeabilization in cell death: concepts and challenges. Mitochondrion 19 Pt A, 49-57
Ross SM, McManus IC, Harrison V, Mason O (2013) Neurobehavioral problems following low-level exposure to organophosphate pesticides: a systematic and meta-analytic review. Crit Rev Toxicol 43:21–44
Roth TL, Nayak D, Atanasijevic T, Koretsky AP, Latour LL, McGavern DB (2014) Transcranial amelioration of inflammation and cell death after brain injury. Nature 505:223–228
Samsel A, Seneff S (2015) Glyphosate, pathways to modern diseases III: manganese, neurological diseases, and associated pathologies. Surg Neurol Int 6:45
Shou L, Bei Y, Song Y, Wang L, Ai L, Yan Q, He W (2019) Nrf2 mediates the protective effect of edaravone after chlorpyrifos-induced nervous system toxicity. Environ Toxicol 34(5):626–633
Slotkin TA (2011) Does early-life exposure to organophosphate insecticides lead to prediabetes and obesity? Reprod Toxicol 31:297–301
Stipanuk MH, Dominy JE Jr, Lee JI, Coloso RM (2006) Mammalian cysteine metabolism: new insights into regulation of cysteine metabolism. J Nutr 136:1652S–1659S
Suke SG, Ahmed RS, Pathak R, Tripathi AK, Banerjee BD (2008) Attenuation of phosphamidon-induced oxidative stress and immune dysfunction in rats treated with N-acetylcysteine. Braz J Med Biol Res 41:765–768
Sun Y, Oberley LW, Li Y (1988) A simple method for clinical assay of superoxide dismutase. Clin Chem 34:497–500
Sun L, Gu L, Wang S, Yuan J, Yang H, Zhu J, Zhang H (2012) N-acetylcysteine protects against apoptosis through modulation of group i metabotropic glutamate receptor activity. PLoS One 7:e32503
Troy CM, Derossi D, Prochiantz A, Greene LA, Shelanski ML (1996) Downregulation of Cu/Zn superoxide dismutase leads to cell death via the nitric oxide-peroxynitrite pathway. J Neurosci 16:253–261
Wang WY, Tan MS, Yu JT, Tan L (2015) Role of pro-inflammatory cytokines released from microglia in Alzheimer's disease. Ann Transl Med 3:136
Yao X, Miao W, Li M, Wang M, Ma J, Wang Y, Miao L, Feng H (2010) Protective effect of albumin on VEGF and brain edema in acute ischemia in rats. Neurosci Lett 472:179–183
Yi JH, Hoover R, McIntosh TK, Hazell AS (2006) Early, transient increase in complexin I and complexin II in the cerebral cortex following traumatic brain injury is attenuated by N-acetylcysteine. J Neurotrauma 23:86–96
Yu ZP, Matsuoka M, Wispriyono B, Iryo Y, Igisu H (2000) Activation of mitogen-activated protein kinases by tributyltin in CCRF-CEM cells: role of intracellular Ca(2+). Toxicol Appl Pharmacol 168:200–207
Zhang P, Zhong S, Wang G, Zhang SY, Chu C, Zeng S, Yan Y, Cheng X, Bao Y, Hocher B, Yang X (2018) N-acetylcysteine suppresses lps-induced pathological angiogenesis. Cell Physiol Biochem 49:2483–2495
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Responsible editor: Philippe Garrigues
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mahmoud, S.M., Abdel Moneim, A.E., Qayed, M.M. et al. Potential role of N-acetylcysteine on chlorpyrifos-induced neurotoxicity in rats. Environ Sci Pollut Res 26, 20731–20741 (2019). https://doi.org/10.1007/s11356-019-05366-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-019-05366-w