
Abstract
This paper describes the stability study performed in seawater and seawater extracts (spiked at ~ 200 ng/L) for 23 emerging contaminants. Four different alternatives were tested at six different times (0, 3, 10, 17, 24 and 31 days): (i) seawater at 4 °C, (ii) mixed-mode solid-phase extraction cartridge (Bond Elute Plexa and Strata X-AW) stored at − 20 °C, (iii) polyethersulfone hollow fibre stored at − 20 °C and (iv) methanol extracts once the samples were extracted from PES hollow fibre and stored at − 20 °C. Moreover, the integrity of the supporting polymeric phases was studied by Raman, optical microscopy, differential scanning calorimetric and thermogravimetric analysis. As may be expected, seawater samples showed the lowest stability (losses between 21 and 99%) while methanol extract provides stable results (losses < 30%) over the tested period. In the case of solid-phase cartridges, the stability profile showed an average loss of 7% while, in polyethersulfone hollow fibres, losses up to 58% were observed. Finally, we were able to relate the lower efficiency of polyethersulfone fibres with the wettability of this material based on the thermogravimetric analysis.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The analysis of the so-called emerging contaminants, i.e. the potentially hazardous compounds that are not under any environmental regulation (Postigo and Barceló 2015), is facing a number of methodological challenges. Among them, we can highlight the design of proper extraction and preconcentration procedures allowing the screening of the widest variety of compounds in a single run, and the strategies to assure the integrity of the samples up to the analysis, since most of the compounds considered are bioactive and their lack of stability can hamper all the analytical efforts (Baker and Kasprzyk-Hordern 2011a; Fedorova et al. 2014; Petrovic 2014).
Regarding the last point, we tend to assume the integrity of the analytes during the sampling step while the storage of the samples should assure their stability, as pointed recently (AMCTB No 65 2015). The increasing interest for the analysis of contaminants of emerging concern, the extended use of passive sampling methods (Miège et al. 2015) and the management requirements when a large number of samples are being processed have opened the discussion about the stability of the samples as well as the best approaches to assure their preservation. In this sense, we can highlight the review for pharmaceuticals in natural waters by Mompelat et al. (2013), the analysis of illicit drugs in sewers and wastewaters by McCall et al. (2016) or the analysis of antibiotics in purified water samples by Llorca et al. (2014).
Concerning the passive sampling methods and its increasing use (Miège et al. 2015), new sampling methodologies such as semipermeable membrane devices (Sultana et al. 2017), Chemcatcher (Kaserzon et al. 2014; Vermeirssen et al. 2012) and polar organic chemical integrative samplers (POCIS) (Iparraguirre et al. 2017; Mijangos et al. 2018) allow the direct analysis from water matrices avoiding, to some extent, the stability issues. However, issues such as the preservation of compounds in different sorbents during the passive sampling may also arise, as pointed by Carlson et al. (2013).
As pointed before, labile analytes such as pharmaceuticals and pesticides are bioactive and hence may undergo different chemical, physical and biological processes from the sampling up to the analysis (Fatta-Kassinos et al. 2011). Thus, depending on stability, quantifying a compound that has been released several hours previously may, in fact, lead to a significant underestimation of the actual amount of residue present. As a consequence, the procedures and strategies to collect and handle the samples are usually guided by the compliance to the existing regulations or to the proper laboratory procedures (Baker and Kasprzyk-Hordern 2011a; Mompelat et al. 2013). Typically, the factors studied are the influence of suspended solids (Baker and Kasprzyk-Hordern 2011a); the addition of a preserving agent (González-Mariño et al. 2010; Llorca et al. 2014); and the storage conditions (temperature, pH and time) (Baker and Kasprzyk-Hordern 2011a; Mompelat et al. 2013).
One of the options is the use of solid-phase extraction (SPE) cartridges (González-Mariño et al. 2010; Petrović and Barceló 2000; Turiel et al. 2004) because we gain the extraction of the analytes and we save a lot of space in the labs. On the other hand, the growing interest for passive samplers such as POCIS (Carlson et al. 2013) can provide simplified procedures to sampling and storing but we lack the knowledge regarding the sorptive features on the different polymers.
Therefore, the aim of this work was the evaluation of the stability of 23 organic contaminants (21 emerging compounds and 2 priority contaminants) during 1 month and under different preservation procedures. For this purpose, four different storage conditions were tested: (i) seawater samples stored at 4 °C, (ii) preconcentration of spiked seawater in a SPE cartridge with a mixture of Bond Elute Plexa and Strata X-AW sorbents (commonly used as passive sampler sorbents (Fauvelle et al. 2012; Kaserzon et al. 2012, Kaserzon et al. 2014; Mijangos et al. 2018) and stored at − 20 °C, (iii) preconcentration in PES hollow fibres (disposable polymeric materials used in microextraction techniques (Bizkarguenaga et al. 2015; Blanco-Zubiaguirre et al. 2014; Mijangos et al. 2017) and stored at − 20 °C and, finally, (iv) the storage of methanol extracts (obtained from seawater preconcentration in PES fibres) at − 20 °C. The target analytes include herbicides, hormones, lifestyle products (stimulants and artificial sweeteners), personal care products, phytoestrogens, industrial chemicals (corrosion inhibitor and perfluoroalkyl substances) and pharmaceuticals (dihydrofolate reductase inhibitor, fluoroquionolones, sulfonamides, dihydrofolate reductase inhibitor (DHFR inhibitor), tricyclic antidepressants, antihypertensives, anti-inflammatories, β-blocker cardiovascular drugs, lipid regulators, angiotensin II receptor antagonists (ARA-IIs) and anticonvulsant psychiatric drug). Additionally, supporting polymeric phases’ (PES, Plexa and Strata X-AW) integrity was also evaluated by means of Raman spectroscopy, optical microscopy and differential scanning calorimetric and thermogravimetrical analysis.
Material and methods
Reagents and materials
The selection of the target pollutants was carried out taking into account their presence and relevance on the environment (Brack et al. 2017; Busch et al. 2016; Tousova et al. 2017). According to these criteria, 23 organic pollutants with urban, rural and industrial use were selected, which cover a wide variety of physicochemical properties as shown in Table 1, including their chemical structure and some physicochemical parameters.
2-Hydroxybenzothiazole (OBT), amitriptyline hydrochloride, butylparaben, caffeine, carbamazepine, lineal perfluorooctane sulfonic acid (PFOS), imipramine hydrochloride, lineal nonafluorobutanesulfonic acid (PFBS), progesterone and sulfadiazine were purchased from Sigma-Aldrich (St. Louis, MO, USA). Atrazine, diuron, norfloxacin hydrochloride, sulfamethoxazole and trimethoprim were acquired from Fluka (Buchs, Switzerland). Acesulfame potassium was supplied by Supelco (Bellefonte, PA, USA), and ketoprofen, bezafibrate and propranolol were acquired from MP biomedicals (Illkirch Cedex, France). Genistein and genistin were purchased from Extrasynthese (Lyon, France), perfluorooctanesulfonamide (PFOSA) from Dr. Ehrenstofer (Augsburg, Germany) and irbesartan from Sanofi (Paris, France). The purity of all the target analytes was higher than 95%.
Individual stock standard solutions were dissolved on a weight basis in methanol (MeOH, UHPLC-MS MeOH, Scharlab, Barcelona, Spain) in order to prepare approximately 1000–2500 mg/L solutions. However, the addition of 100 μL sodium hydroxide 1 M (NaOH, 98%, Panreac, Barcelona, Spain) was necessary for the proper dissolution of fluoroquinolone antibiotics as described by Gros et al. (2013). One hundred milligrams per litre dilutions were prepared in MeOH every month, and dilutions at lower concentrations containing all analytes were prepared daily in MeOH: Milli-Q (30:70, v:v). All the chemical standard solutions were stored at − 20 °C.
The most relevant characteristics and suppliers of the polymers evaluated in the present work are listed in Table 2. Empty SPE tubes (6 mL) and polypropylene (PP) frites were purchased from Supelco. PES hollow fibre agitation was carried out using 150-mL glass vessels provided by ServiQuimia (Tarragona, Spain) in a 15-position magnetic stirrer (Gerstel, Mülheim an der Ruhr, Germany). Desorption was made in 1.5-mL Eppendorf tubes purchased from Eppendorf (Berzdorf, Germany) using a Digital Ultrasonic Cleaner (2500 mL, USB Axtor by Lovango, Barcelona, Spain). Ethylenediaminetetraacetic acid sodium salt (Na2EDTA, 99.0–101.1%, Panreac), formic acid (HCOOH ≥ 98%, Scharlau, Barcelona, Spain), ammonia (25% as NH3, Panreac) and sodium chloride (NaCl, > 99.8%, Merck) were used for matrix modification and elution step. MeOH (HPLC grade, 99.9%) was supplied by LabScan (Dublin, Ireland).
The extracts were evaporated using a Turbovap LV Evaporator (Zymark, Hopkinton, USA) under a gentle stream of nitrogen (> 99.999% of purity) supplied by Messer (Tarragona, Spain). The extracts were filtered through PP filters (0.22 μm, 13 mm, Phenomenex, California, USA). Milli-Q (< 0.05 μS/cm, Milli-Q, Millipore) water and UHPLC-MS MeOH (Optima, Scharlau, Barcelona, Spain) were used as mobile phase eluents and HCOOH (Optima, Fischer Scientific, Geel, Belgium) for mobile phase modification. High-purity nitrogen gas (> 99.999%) supplied by Messer was used as collision gas, and nitrogen gas (99.999%) provided by AIR Liquid (Madrid, Spain) was used as both nebuliser and drying gas.
Stability tests
The removal of suspended particulates from water samples may avoid further degradation or losses of analytes through their adsorption onto the solid particles, but the retention of these analytes in the filters should be also thoroughly considered (Baker and Kasprzyk-Hordern 2011a; Petrovic 2014). In this sense, unfiltered seawater samples were collected in 2-L clear-PP bottles as previously described by Llorca et al. (2014) from the Plentzia Marine Station (PiE-UPV/EHU, Basque Country, Northern Spain) and used for the stability experiments. Furthermore, some physicochemical parameters (temperature, dissolved oxygen, pH, redox potential, conductivity and salinity, see Table S1 in online resource) of the seawater were monitored with a multiparametric probe (EXO 2, YSI, USA).
The experiments were carried out spiking the seawater (100 mL) with a mixture of the 23 analytes at a final concentration of ~ 200 ng/L each one. In parallel to the spiked samples, a non-spiked control sample (blank) was also processed in duplicate alongside each of the stability tests and at the same conditions. The experiments were performed in triplicate for each preservation mode at 6 different sampling times (after 0, 3, 10, 17, 24 and 31 days), and all the samples were analysed at the same day by LC–MS/MS (Mijangos et al. 2017) (see “Liquid chromatography–tandem mass spectrometry analysis”).
In the case of preservation mode (i), 100 mL of unfiltered seawater were stored at 4 °C in the pre-cleaned PP bottles. In the case of mode (ii), 200 mg SPE cartridges (a 1:1 mixture of Strata X-AW and Plexa, as the sorbent composition used for passive sampler previously published (Mijangos et al. 2018)) were prepared from 100 mL spiked seawater samples. SPE cartridges were sequentially conditioned with 5 mL of MeOH and 5 mL of ultrapure water. Then, the water sample (100 mL) was percolated through the cartridge assisted by a vacuum pump at ca. 5 mL/min.
In the case of mode (iii), 4 pre-cleaned PES hollow fibres of 4 cm (final weight of approx. 50 mg) were used, according to the previously published work (Mijangos et al. 2017). First of all, the fibres were cut using a sharp blade and conditioned by soaking overnight in MeOH and air dried. Two aliquots of 120 mL of spiked seawater (dual extraction) were directly poured into 150-mL extraction vessels, and NaCl and Na2EDTA were added to achieve final concentrations of 30% (w/v) and 0.1% (w/w), respectively. The pH of each aliquot was fixed at pH = 2 and pH = 10, and finally, hollow fibres and a magnetic stirrer were also added. Thereafter, vessels were closed and extraction was performed at room temperature (RT) and at 800 rpm overnight (14 h). Once the sorption step was over, the polymers were removed and rinsed with Milli-Q water in order to eliminate salt residues, and, finally, dried with a clean tissue and stored in an air-tight freezer bag at − 20 °C.
Finally, in the case of mode (iv), PES fibres were used following the extraction procedure described before, and once the extraction was accomplished, the fibres were introduced into a 1.5-mL Eppendorf tube containing 1 mL of MeOH and soaked for 32 min in an ultrasound bath and then, the methanolic extracts were stored in Eppendorf tubes at − 20 °C.
To run the analysis (18 aliquots per preservation mode), water samples (mode (i)) were extracted by SPE cartridges according to the previously published work (Mijangos et al. 2017). In the case of mode (ii), the cartridges were washed with 6 mL of ultrapure water, vacuum dried and eluted with 6 mL of 2.5% (v/v) ammonia solution in methanol followed by 6 mL of methanol (Mijangos et al. 2018). PES fibres used in modes (iii) and (iv) were extracted using the procedure described before. All the extraction solutions from modes (i–iv) were always evaporated to dryness under a gentle stream of nitrogen at 35 °C and reconstituted in 200 μL of MeOH:Milli-Q (30:70, v:v). Finally, the reconstituted extracts were filtered through a 0.2 μm PP filter before the LC–MS/MS analysis.
The target analyte stability was calculated according to Eq. (1), and a 100% result represents a lack of analyte losses or degradation:
where \( {A}_{x,\mathrm{sp}}^i \) and \( {A}_{x,\mathrm{nsp}}^i \)correspond to the chromatographic peak areas of analyte x from the spiked (sp) and non-spiked (nsp) samples, respectively, at time i, and \( {A}_{x,\mathrm{sp}}^0 \) and \( {A}_{x,\mathrm{nsp}}^0 \) are the corresponding peak areas at day 0. A significant (mean) loss of 30% in the recovery of the analytes was chosen to point out the lack of stability during a given storage preservation mode and time, since the precision attributable to an analytical method, expressed as RSD (inter-day precision), must be ≤ 30% according to the European Commission decision 2002/657/EC (Commission E 2002).
Characterisation of sorptive materials
PES, Plexa and Strata X-AW were individually examined prior to and after storage at − 20 °C and RT for a month. The surface and the cross section of the polymer materials were examined by a Nikon SMZ800 stereomicroscope coupled to a NIKON DS-RI1 at × 40 magnifications. Chemical characterisation of the sorptive materials was assured by means of Raman spectroscopy. They were analysed using a portable Renishaw RA 100 Raman spectrometer (Renishaw, Gloucestershire, UK) using either the 785 nm or the 514 nm excitation laser. Measured scans were accumulated during 50 s at 100% of the maximum power of the used laser. The homogeneity of the PES hollow fibre was tested by acquiring longitudinally ten Raman spectra per fibre (one measurement per 1.5 mm). The software used to collect and process the Raman spectra was BWspec4 and Omnic (Nicolet, Madison, WI, USA).
The wettability and thermal stability of the polymeric materials were studied by differential scanning calorimeter (DSC) by a Mettler Toledo Differential Scanning Calorimeter instrument (model DSC822). Ten milligrams of each polymeric material was subjected to 5 sequential heating/cooling cycles: the first 4 were done consecutively and the 5th run was performed after having the polymer 1 h out of the measuring chamber (nitrogen ambient). Temperature range was from 0 to 200 °C, and the scanning rate was of 20 °C/min. Furthermore, a thermogravimetric analysis (TGA) was performed in a Mettler Toledo TGA/SDTA 851 system. Ten milligrams of solid-phase samples was kept during 30 min at 20 °C prior to the measurement and then heated from 20 to 800 °C. The scanning rate was 10 °C/min, and all measurements were carried out under nitrogen atmosphere.
Liquid chromatography–tandem mass spectrometry analysis
An Agilent 1260 series HPLC chromatograph equipped with a degasser, binary pump, autosampler and a column oven coupled to an Agilent 6430 triple quadrupole (QqQ) mass spectrometer with electrospray ionisation (ESI) source (Palo Alto, CA, USA) was employed. For analyte separation, a Kinetex F5 100 Å core-shell (2.1 mm × 100 mm, 2.6 μm) column coupled to a Kinetex F5 pre-column (2.1 mm × 4.6 mm, 2.6 μm from Phenomenex (Torrance, CA, USA) was used. The column temperature and the injection volume were set to 35 °C and 5 μL, respectively. The separation of the target analytes was carried out at a flow rate of 0.3 mL/min. Under optimised conditions [25], a binary mixture consisting of water:MeOH (95:5, v:v) (phase A) and MeOH:water (95:5, v:v) (phase B), both containing 0.1% of HCOOH, was used for gradient separation of target analytes. The gradient profile started with 30% B, and it was increased to 50% in 4 min and maintained for 12 min. Then, it was increased to 90% B and it was kept constant for 10 min. Initial gradient conditions (30% B) were then recovered in 6 min and held constant for another 10 min (post-run step). ESI was carried out using a N2 flow rate of 12 L/min, a capillary voltage of 3500 V, a nebuliser pressure of 45 psi and a source temperature of 350 °C. Quantification was performed in the selected reaction monitoring (SRM) acquisition mode by recording the two most intense transitions for each analyte (the most sensitive was chosen as the quantifier and the second one as qualifier) when it was possible. Both, negative and positive voltages, according to the target analytes, were simultaneously applied in a single injection. Optimum parameter values for each target compound are summarised in Table S2 in Online Resource. Instrumental operations, data acquisition and peak integration were performed with the MassHunter Workstation Software (v B.06.00, Agilent Technologies).
Results and discussions
Quality control
Some physicochemical parameters (temperature, dissolved oxygen, pH, redox potential, conductivity and salinity) of the seawater were measured along the experiment (see Table S1 in Online Resource). Temperature, pH and salinity were constant with average values of 13.6 ± 0.2 °C, 0.4 ± 0.2 and 33.7 ± 0.2 psμ, respectively, and the redox potential values (144 ± 25 mV) showed an RSD of 18%.
Furthermore, the analytical figures of merit in real spiked seawater samples (n = 3, 100 ng/L) of both the previously published PES-LC-MS/MS (Mijangos et al. 2017) methodology and in the case of SPE-LC-MS/MS method are summarised in Table S2 in Online Resource. The quantification of the target analytes in real seawater was carried out using an external calibration together with surrogate corrections approach for SPE, while, in the case of PES method, a procedural calibration with Milli-Q using isotopically labelled analogues as surrogates was used. In this sense, process efficiency, apparent recovery (recovery after correction with the corresponding surrogate included in Table S2) and method quantification limits (MQLs) were determined. MQLs were calculated using the Eq. (2) (Baker and Kasprzyk-Hordern 2011b; Huntscha et al. 2012; Kasprzyk-Hordern et al. 2008).
where LOQ (ng/mL) is the instrumental quantification limit (included in Table S2 in Online Resource), PE (%) is the process efficiency of the analyte in the corresponding matrix (see Table S2) and CF is the analyte concentration factor according to the developed procedures.
Additionally, during the sample treatment, control samples (samples spiked at known concentration level, n = 3) and procedural blanks (n = 3) were analysed periodically every 12–15 samples. RSDs in the range of 3–30% were obtained for all the analytes, and concentrations lower than their MQLs were obtained in the case of blanks for the target compounds.
Characterisation of sorptive materials
The polymers used in the present study (Plexa, Strata X-AW and PES) were characterised chemically and thermally before and after being stored at − 20 °C and RT for a month. Good-quality Raman spectra (see Fig. S1 for the PES hollow fibre as example) and microscopy surface images (see Fig. S2a-l) were obtained for all the sorptive phases before and after storing, and no differences were observed at the two temperatures.
However, when the thermal degradation was studied by running a TGA curve, in the case of PES hollow fibre, the thermal characteristics obtained from TGA and first-derived thermogravimetric (DTG) curves (seen in Fig. 1a, b) showed difference between the polymers without storage and after low-temperature storage. When the fibres were kept at RT, a significant weight loss temperature was observed at 550 °C and attributed to the decomposition of polymer main chain (Cao et al. 2011; Guan et al. 2005; Sharma and Bijwe 2012). When PES fibres were kept at − 20 °C for 1 month, an additional sharp weight (35% of the total mass) can be seen at 100 °C and this loss can be related to the desorption of water. These results are in total agreement with published data (Cao et al. 2011; Guan et al. 2005; Sharma and Bijwe 2012) where it was observed that water can be bonded through the sulfonic groups of polyethersulfone polymers. Plexa and Strata X-AW did not show any significant changes (see Fig. 1c–d) in thermal behaviour; the weight loss origin from water content was <5% in both cases.

Thermogravimetric (TGA) curve (left axis, line) and first derivate thermogravimetric (DTG, right axis, dots) of the studied polymers before and after storage at − 20 °C for 1 month: a PES hollow fibre before storage; b PES hollow fibre after storage; c PLEXA after storage; d Strata X-AW after storage
Finally, the wettability of these polymers was studied by running DSC analysis in a sequential way. As shown in Fig. 2a, PES hollow fibres showed two signals in its thermogram: a broad peak around 100 °C, due to the desorption of water molecules present in the polymers (Cao et al. 2011; Sharma and Bijwe 2012) and a glass transition temperature (Tg) at 230 °C, which is in agreement with the values reported in the literature for pure PES (Bolong et al. 2009; Cao et al. 2011; Prieto et al. 2012; Sharma and Bijwe 2012). Regarding the wettability, the removal of water content of the PES fibre was achieved after running the scan several times (runs 1–4 in Fig. 2a) since the humidity peak was significantly smoothed at every scan. Furthermore, the observed increase of the glass transition temperature is a consequence of the plastification induced by the humidity that lowers Tg. Finally, once the fibre was released from the inert gas chamber of the DSC for an hour (run 5 in Fig. 2a), the broad peak corresponding to the humidity increased again suggesting that the PES hollow fibre can re-adsorb water. Thus, PES hollow fibre has the ability to re-uptake water from the air even after being totally dried (Guan et al. 2005). On the contrary, the signals of Plexa and Strata X-AW (see Fig. 2 b and c, respectively) remain constant after getting dried. These results suggest that the polymers chosen (PLEXA, Strata X-AW and PES hollow fibre) have a good thermal and chemical stability; however, the hydrophobicity of the PES hollow fibre, closely linked to the chemical structure of the polymer, may be an issue.

Sequential differential scanning calorimetric (DSC) carried out for the three studied polymers after storing at − 20 °C for 1 month: a PES hollow fibre, b Plexa and c Strata X-AW. First four measurements (continuous line) were run sequentially (heating/cooling cycles) but the 5th run (dots) was performed after having each polymer 1 h out of the measuring chamber (nitrogen inert gas ambient)
Stability test
The variation of the concentrations of all the analytes along the storage time in the four modes studied in this work is shown in Fig. 3a–d. As mentioned before (see “Characterisation of sorptive materials”), the storage procedure assures the stability when the losses along the storage time are below 30%.

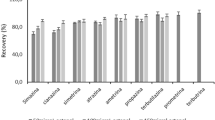
Relative recovery percentage of each analyte at 6 times (0, 3, 10, 17, 24 and 31 days) preserved at a raw seawater at 4 °C, b SPE cartridges at − 20 °C, c PES hollow fibres stored at − 20 °C and d 100% methanol extracts stored at − 20 °C
In the case of procedure (i), three profiles were observed, as shown in Fig. 3a: a declining profile (78% of the studied analytes), an increasing profile and constant concentrations throughout the experimental period. After 31 days, statistically significant losses (within 20–45% at a p level < 0.05 in the analysis of variance) were observed for atrazine, bezafibrate, butylparaben, caffeine, diuron, ketoprofen, norfloxacin, OBT, propranolol, sulfadiazine, sulfamethoxazole and trimethoprim, whereas amitriptyline, imipramine, genistein, genistin, irbesartan and progesterone reduced quantitatively (> 99%) their initial concentrations in just 3 days. These behaviours could be attributed to the chemical structure and reactivity of the studied analytes. With regard to pharmaceutical-like compounds, numerous studies have reported the lack of stability in aqueous samples (Baker and Kasprzyk-Hordern 2011a; Fedorova et al. 2014; Llorca et al. 2014; Mompelat et al. 2013). Baker et al. (Baker and Kasprzyk-Hordern 2011a) described a thorough verification of methodologies commonly used for the storage of aqueous samples and for the analysis of pharmaceuticals and illicit drugs, and observed that antidepressant showed a poor stability with a recovery decreased of 61% after 72 h in unfiltered wastewater samples. Turiel et al. (2004) studied the degradation of fluoroquinolones under different storage conditions (time, light and temperature) for 2 weeks, and the analyte losses were mainly attributed to photolysis (after 2 weeks, a loss of 50% of the initial concentration was observed).
The increasing profile (up to 146%) was detected in the case of PFOS accompanied by a parallel signal decrease (up to 42%) of its parent compound PFOSA. Similar degradation pathway of PFOSA precursor into the stable PFOS end-product has been reported in the literature (Buck et al. 2011; Zhang et al. 2017). Finally, only acesulfame, carbamazepine and PFBS remain constant during the 31-day evaluation (p level > 0.05, according to the analysis of variance, ANOVA). These results are in good agreement with those of Van Stempvoort et al. (2011), which compared refrigerated and frozen environmental samples for the stability of artificial sweeteners (acesulfame, cyclamate, saccharin, sucralose) over a storage time of 13 months and found acesulfame was stable during this period. Due to their high stability in aquatic media, acesulfame and carbamazepine compounds have been proposed as tracers of human wastewater contamination in environmental samples (Huntscha et al. 2012; Jekel et al. 2015; Lange et al. 2012; Mawhinney et al. 2011).
In the case of the SPE cartridges, the average loss of all compounds after 31 days of storage was 7% with a maximum loss of 24% for OBT (see Fig. 3b). Therefore, the short-term preservation of extracted samples in SPE cartridges in the freezer (− 20 °C) is a good approach. The advantages of using SPE cartridges for these purposes have been previously described in several works (Baker and Kasprzyk-Hordern 2011a; Fedorova et al. 2014; Mompelat et al. 2013).
On the contrary, though a close stability pattern would have been expected in PES hollow fibres, the stability profiles obtained in PES were quite different from those obtained onto SPE cartridges (see Fig. 3 b and c for SPE and PES, respectively). PES hollow fibres showed remarkable losses in analyte concentrations after 31 days for acesulfame (45% remaining after 31 days), caffeine (65%), genistein (42%), genistin (60%), norfloxacin (50%), OBT (45%), PFBS (65%), sulfadiazine (43%) and sulfamethoxazole (53%). In contrast to the well-known stability onto SPE cartridges (C18 and/or HLB) (Llorca et al. 2014; McCall et al. 2016; Mompelat et al. 2013), there is no published data on stability tests for PES polymer material, even though it is highly used in POCIS as the supporting membrane (Carlson et al. 2013; Posada-Ureta et al. 2017; Vermeirssen et al. 2012) and in sorptive microextraction methods (Bizkarguenaga et al. 2015; Blanco-Zubiaguirre et al. 2014; Prieto et al. 2012; Ros et al. 2015).
Finally, as can be seen in Fig. 3d, all the analyte concentrations (< 7 0%) remain stable up to 31 days in the MeOH extracts allowing the accurate estimation of the concentrations of the target compounds in environmental matrices.
Regarding the four different modes evaluated and in the case of some of the studied compounds, only carbamazepine remained constant regardless of the preservation mode after 31 days. Remarkable losses onto PES hollow fibres were observed in compounds such as acesulfame (55%) and PFBS (35%) that showed a high stability in water (91–108% and 87–102%, respectively). The stability of the phytoestrogens, OBT, fluoroquinolones and sulphonamides was rather low onto PES hollow fibres (42–60% remaining concentrations after 31 days) as well as in seawater. In contrast, amitriptyline, butylparaben, imipramine, irbesartan, progesterone and PFOSA were significantly more stable onto PES hollow fibres (losses < 20%) compared with seawater (losses up to 99%).
The patterns observed in the PES hollow fibre might not be related to the degradation of those compounds in the polymer but to the presence of the low amount of water observed in the previous section that may help to solubilise and to lose some analytes such as genistein, genistin, OBT, PFOSA, sulfadiazine or sulfamethoxazole as it happens to a similar extent in water (see the solubility values collected in Table 1).
Conclusions
According to the results obtained in this work, the best way to assure the stability of the water samples containing polar or slightly polar emerging contaminants is either to keep the MeOH extracts after being extracted the samples by SPE or any other procedure or to keep the extracted samples in SPE cartridges. Both procedures assure a high recovery of a wide amount of contaminants typically found in aquatic media for a short-term period. This way, the management of the sample analysis can be effectively carried out. Furthermore, PLEXA, Strata X-AW and PES hollow fibre showed a good thermal and chemical stability to be used as potential solid phases but the wettability of the PES fibres has been linked to the lack of stability of a number of compounds. A deeper study of the polymeric materials showed that the losses observed in PES hollow fibres were related to the capability of the polymer to re-absorb water, which might degrade biotically some analytes or redissolve them due to their high water solubility.
References
AMCTB No 65 (2015) Sample stability studies for environmental analysis. Analytical Methods 7 (6):2256-2257
Baker DR, Kasprzyk-Hordern B (2011a) Critical evaluation of methodology commonly used in sample collection, storage and preparation for the analysis of pharmaceuticals and illicit drugs in surface water and wastewater by solid phase extraction and liquid chromatography–mass spectrometry. J Chromatogr A 1218:8036–8059
Baker DR, Kasprzyk-Hordern B (2011b) Multi-residue analysis of drugs of abuse in wastewater and surface water by solid-phase extraction and liquid chromatography–positive electrospray ionisation tandem mass spectrometry. J Chromatogr A 1218:1620–1631
Bizkarguenaga E, Zabaleta I, Iparraguirre A, Aguirre J, Fernández LÁ, Berger U, Prieto A, Zuloaga O (2015) Enrichment of perfluorinated alkyl substances on polyethersulfone using 1-methylpyperidine as ion-pair reagent for the clean-up of carrot and amended soil extracts. Talanta 143:263–270
Blanco-Zubiaguirre L, Delgado A, Ros O, Posada-Ureta O, Vallejo A, Prieto A, Olivares M, Etxebarria N (2014) Assessment of commercially available polymeric materials for sorptive microextraction of priority and emerging nonpolar organic pollutants in environmental water samples. Environ Sci Pollut Res 21:11867–11883
Bolong N, Ismail AF, Salim MR, Rana D, Matsuura T (2009) Development and characterization of novel charged surface modification macromolecule to polyethersulfone hollow fiber membrane with polyvinylpyrrolidone and water. J Membr Sci 331:40–49
Brack W, Dulio V, Ågerstrand M, Allan I, Altenburger R, Brinkmann M, Bunke D, Burgess RM, Cousins I, Escher BI, Hernández FJ, Hewitt LM, Hilscherová K, Hollender J, Hollert H, Kase R, Klauer B, Lindim C, Herráez DL, Miège C, Munthe J, O’Toole S, Posthuma L, Rüdel H, Schäfer RB, Sengl M, Smedes F, van de Meent D, van den Brink PJ, van Gils J, van Wezel AP, Vethaak AD, Vermeirssen E, von der Ohe PC, Vrana B (2017) Towards the review of the European Union Water Framework Directive: recommendations for more efficient assessment and management of chemical contamination in European surface water resources. Sci Total Environ 576:720–737
Buck RC, Franklin J, Berger U, Conder JM, Cousins IT, de Voogt P, Jensen AA, Kannan K, Mabury SA, van Leeuwen SP (2011) Perfluoroalkyl and polyfluoroalkyl substances in the environment: terminology, classification, and origins. Integr Environ Assess Manag 7:513–541
Busch W, Schmidt S, Kühne R, Schulze T, Krauss M, Altenburger R (2016) Micropollutants in European rivers: a mode of action survey to support the development of effect-based tools for water monitoring. Environ Toxicol Chem 35:1887–1899
Cao XL, Cheng C, Yin ZH, Bai PL, Wei Q, Fang BH, Zhao CS (2011) Synthesis, characterization, and application of polyethersulfone bound-iminodiacetic acid. J Appl Polym Sci 120:345–350
Carlson JC, Challis JK, Hanson ML, Wong CS (2013) Stability of pharmaceuticals and other polar organic compounds stored on polar organic chemical integrative samplers and solid-phase extraction cartridges. Environ Toxicol Chem 32:337–344
Commission E (2002) Commission Decision (EC) 2002/657/EC of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Parliament and of the council. Off J Eur Union:L221/8–L221/10
Fatta-Kassinos D, Vasquez MI, Kümmerer K (2011) Transformation products of pharmaceuticals in surface waters and wastewater formed during photolysis and advanced oxidation processes – degradation, elucidation of byproducts and assessment of their biological potency. Chemosphere 85:693–709
Fauvelle V, Mazzella N, Delmas F, Madarassou K, Eon M, Budzinski H (2012) Use of mixed-mode ion exchange sorbent for the passive sampling of organic acids by polar organic chemical integrative sampler (POCIS). Environ Sci Technol 46:13344–13353
Fedorova G, Golovko O, Randak T, Grabic R (2014) Storage effect on the analysis of pharmaceuticals and personal care products in wastewater. Chemosphere 111:55–60
González-Mariño I, Quintana JB, Rodríguez I, Cela R (2010) Determination of drugs of abuse in water by solid-phase extraction, derivatisation and gas chromatography–ion trap-tandem mass spectrometry. J Chromatogr A 1217:1748–1760
Gros M, Rodríguez-Mozaz S, Barceló D (2013) Rapid analysis of multiclass antibiotic residues and some of their metabolites in hospital, urban wastewater and river water by ultra-high-performance liquid chromatography coupled to quadrupole-linear ion trap tandem mass spectrometry. J Chromatogr A 1292:173–188
Guan R, Zou H, Lu D, Gong C, Liu Y (2005) Polyethersulfone sulfonated by chlorosulfonic acid and its membrane characteristics. Eur Polym J 41:1554–1560
Huntscha S, Singer HP, McArdell CS, Frank CE, Hollender J (2012) Multiresidue analysis of 88 polar organic micropollutants in ground, surface and wastewater using online mixed-bed multilayer solid-phase extraction coupled to high performance liquid chromatography–tandem mass spectrometry. J Chromatogr A 1268:74–83
Iparraguirre A, Prieto A, Vallejo A, Moeder M, Zuloaga O, Etxebarria N, Paschke A (2017) Tetraphasic polar organic chemical integrative sampler for the determination of a wide polarity range organic pollutants in water. The use of performance reference compounds and in-situ calibration. Talanta 164:314–322
Jekel M, Dott W, Bergmann A, Dünnbier U, Gnirß R, Haist-Gulde B, Hamscher G, Letzel M, Licha T, Lyko S, Miehe U, Sacher F, Scheurer M, Schmidt CK, Reemtsma T, Ruhl AS (2015) Selection of organic process and source indicator substances for the anthropogenically influenced water cycle. Chemosphere 125:155–167
Kaserzon SL, Kennedy K, Hawker DW, Thompson J, Carter S, Roach AC, Booij K, Mueller JF (2012) Development and calibration of a passive sampler for perfluorinated alkyl carboxylates and sulfonates in water. Environ Sci Technol 46:4985–4993
Kaserzon SL, Hawker DW, Kennedy K, Bartkow M, Carter S, Booij K, Mueller JF (2014) Characterisation and comparison of the uptake of ionizable and polar pesticides, pharmaceuticals and personal care products by POCIS and Chemcatchers. Environ Sci: Processes Impacts 16:2517–2526
Kasprzyk-Hordern B, Dinsdale RM, Guwy AJ (2008) Multiresidue methods for the analysis of pharmaceuticals, personal care products and illicit drugs in surface water and wastewater by solid-phase extraction and ultra performance liquid chromatography–electrospray tandem mass spectrometry. Anal Bioanal Chem 391:1293–1308
Lange FT, Scheurer M, Brauch H-J (2012) Artificial sweeteners—a recently recognized class of emerging environmental contaminants: a review. Anal Bioanal Chem 403:2503–2518
Llorca M, Gros M, Rodríguez-Mozaz S, Barceló D (2014) Sample preservation for the analysis of antibiotics in water. J Chromatogr A 1369:43–51
Mawhinney DB, Young RB, Vanderford BJ, Borch T, Snyder SA (2011) Artificial sweetener sucralose in U.S. drinking water systems. Environ Sci Technol 45:8716–8722
McCall A-K, Bade R, Kinyua J, Lai FY, Thai PK, Covaci A, Bijlsma L, van Nuijs ALN, Ort C (2016) Critical review on the stability of illicit drugs in sewers and wastewater samples. Water Res 88:933–947
Miège C, Mazzella N, Allan I, Dulio V, Smedes F, Tixier C, Vermeirssen E, Brant J, O’Toole S, Budzinski H, Ghestem J-P, Staub P-F, Lardy-Fontan S, Gonzalez J-L, Coquery M, Vrana B (2015) Position paper on passive sampling techniques for the monitoring of contaminants in the aquatic environment – achievements to date and perspectives. Trends Environ Anal Chem 8:20–26
Mijangos L, Ziarrusta H, Olivares M, Zuloaga O, Möder M, Etxebarria N, Prieto A (2017) Simultaneous determination of 41 multiclass organic pollutants in environmental waters by means of polyethersulfone microextraction followed by liquid chromatography–tandem mass spectrometry. Anal Bioanal Chem 410:615–632
Mijangos L, Ziarrusta H, Prieto A, Zugazua O, Olivares M, Usobiaga A, Paschke A, Etxebarria N (2018) Evaluation of polar organic chemical integrative and hollow fibre samplers for the determination of a wide variety of organic polar compounds in seawater. Talanta 185:469–476
Mompelat S, Jaffrezic A, Jardé E, Le Bot B (2013) Storage of natural water samples and preservation techniques for pharmaceutical quantification. Talanta 109:31–45
Petrovic M (2014) Methodological challenges of multi-residue analysis of pharmaceuticals in environmental samples. Trends Environ Anal Chem 1:e25–e33
Petrović M, Barceló D (2000) The stability of non-ionic surfactants and linear alkylbenzene sulfonates in a water matrix and on solid-phase extraction cartridges. Fresenius J Anal Chem 368:676–683
Posada-Ureta O, Olivares M, Delgado A, Prieto A, Vallejo A, Irazola M, Paschke A, Etxebarria N (2017) Applicability of polydimethylsiloxane (PDMS) and polyethersulfone (PES) as passive samplers of more hydrophobic organic compounds in intertidal estuarine environments. Sci Total Environ 578:392–398
Postigo C, Barceló D (2015) Synthetic organic compounds and their transformation products in groundwater: occurrence, fate and mitigation. Sci Total Environ 503–504:32–47
Prieto A, Rodil R, Quintana JB, Rodríguez I, Cela R, Möder M (2012) Evaluation of low-cost disposable polymeric materials for sorptive extraction of organic pollutants in water samples. Anal Chim Acta 716:119–127
Ros O, Vallejo A, Blanco-Zubiaguirre L, Olivares M, Delgado A, Etxebarria N, Prieto A (2015) Microextraction with polyethersulfone for bisphenol-A, alkylphenols and hormones determination in water samples by means of gas chromatography–mass spectrometry and liquid chromatography–tandem mass spectrometry analysis. Talanta 134:247–255
Sharma M, Bijwe J (2012) Influence of molecular weight on performance properties of polyethersulphone and its composites with carbon fabric. Wear 274–275:388–394
Sultana T, Murray C, Hoque ME, Metcalfe CD (2017) Monitoring contaminants of emerging concern from tertiary wastewater treatment plants using passive sampling modelled with performance reference compounds. Environ Monit Assess 189:1–19
Tousova Z, Oswald P, Slobodnik J, Blaha L, Muz M, Hu M, Brack W, Krauss M, Di Paolo C, Tarcai Z, Seiler T-B, Hollert H, Koprivica S, Ahel M, Schollée JE, Hollender J, Suter MJ-F, Hidasi AO, Schirmer K, Sonavane M, Ait-Aissa S, Creusot N, Brion F, Froment J, Almeida AC, Thomas K, Tollefsen KE, Tufi S, Ouyang X, Leonards P, Lamoree M, Torrens VO, Kolkman A, Schriks M, Spirhanzlova P, Tindall A, Schulze T (2017) European demonstration program on the effect-based and chemical identification and monitoring of organic pollutants in European surface waters. Sci Total Environ 601:1849–1868
Turiel E, Martín-Esteban A, Bordin G, Rodríguez AR (2004) Stability of fluoroquinolone antibiotics in river water samples and in octadecyl silica solid-phase extraction cartridges. Anal Bioanal Chem 380:123–128
Van Stempvoort DR, Roy JW, Brown SJ, Bickerton G (2011) Artificial sweeteners as potential tracers in groundwater in urban environments. J Hydrol 401:126–133
Vermeirssen ELM, Dietschweiler C, Escher BI, van der Voet J, Hollender J (2012) Transfer kinetics of polar organic compounds over polyethersulfone membranes in the passive samplers POCIS and Chemcatcher. Environ Sci Technol 46:6759–6766
Zhang L, Lee LS, Niu J, Liu J (2017) Kinetic analysis of aerobic biotransformation pathways of a perfluorooctane sulfonate (PFOS) precursor in distinctly different soils. Environ Pollut 229:159–167
Funding
This work was financially supported by the Ministry of Economy and Competitiveness and the European Regional Development Fund (ERDF) through the project CTM2014-56628-C3-1-R and by the Basque Government through the project IT-742-13. L. Mijangos and H. Ziarrusta are grateful to the Basque Government and to the Spanish Ministry, respectively, for their pre-doctoral fellowships.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Responsible editor: Roland Peter Kallenborn
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 4324 kb)
Rights and permissions
About this article

Cite this article
Mijangos, L., Urain, O., Ruiz-Rubio, L. et al. Short-term stability assessment for the analysis of emerging contaminants in seawater. Environ Sci Pollut Res 26, 23861–23872 (2019). https://doi.org/10.1007/s11356-019-05172-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-019-05172-4