
Abstract
nirK and nirS genes are important functional genes involved in the denitrification pathway. Recent studies about these two denitrifying genes are focusing on sediment and wastewater microbe. In this study, we conducted a comparative analysis of the abundance and diversity of denitrifiers in the epiphyton of submerged macrophytes Potamogeton malaianus and Ceratophyllum demersum as well as in bacterioplankton in the shallow fresh lake Taihu, China. Results showed that nirK and nirS genes had significant different niches in epiphyton and bacterioplankton. Bacterioplankton showed greater abundance of nirK gene in terms of copy numbers and lower abundance of nirS gene. Significant difference in the abundance of nirK and nirS genes also existed between the epiphyton from different submerged macrophytes. Similar community diversity yet different community abundance was observed between epiphytic bacteria and bacterioplankton. No apparent seasonal variation was found either in epiphytic bacteria or bacterioplankton; however, environmental parameters seemed to have direct relevancy with nirK and nirS genes. Our study suggested that submerged macrophytes have greater influence than seasonal parameters in shaping the presence and abundance of bacterial denitrifiers. Further investigation needs to focus on the potential contact and relative contribution between denitrifiers and environmental factors.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Denitrification is an oxygen-sensitive process that transforms significant nitrogen oxide compounds (NO3 − and NO2 −) into gaseous nitrogen (NO, N2O, and N2). There are four enzymes regulating the transformation processes: nitrate reductase, nitrite reductase, nitric oxide reductase, and nitrous oxide reductase. In bacteria, nitrite reductase containing either copper (nirK) or heme cd 1 - (nirS) is the key enzymes to reduce nitrite (NO2−) into nitric oxide (NO) (Antonyuk et al. 2005). Approximately 130 species of denitrifying bacteria allocated in over 50 genera have been documented (You 2005). They are specific bacteria possessing one of the two nitrite reductase enzymes that are regulated by nirK and nirS genes (Zumft 1997). Hence, these two genes become the most frequently used gene markers for tracking denitrifying bacteria (Braker et al. 1998, 2000). Recent studies are focusing on the abundance and population diversity of nir gene in ocean (Bowen et al. 2014; Kraft et al. 2014), sediment (Abell et al. 2010; Hou et al. 2013; Zhang et al. 2015), soil (Attard et al. 2011; Barta et al. 2010; Yuan et al. 2012), and wastewater (Wang et al. 2014). The distribution patterns of distinct species of nir gene-bearing bacteria depend on environmental parameters like crop type (Smith and Ogram 2008), fertilizer type and use level (Enwall et al. 2005; Wolsing and Prieme 2004), land use (Attard et al. 2011; Bremer et al. 2007; Hai et al. 2009), temperature (Braker et al. 2010; Szukics et al. 2010), soil moisture content (Szukics et al. 2010), pH (Braker et al. 2010; Szukics et al. 2010), etc. Physiological differences also affect the distribution of nir gene, for example nirK gene-bearing bacteria is more widespread and more sensitive to environmental variation than nirS gene-bearing bacteria (Smith and Ogram 2008). However, little attention has been paid to the abundance and population diversity of nir gene-bearing bacteria in freshwater lakes or to the epiphyton attached on submerged macrophytes.
Submerged macrophytes are fundamental component in lacustrine ecosystems. They influence the dynamics and processes of the entire ecosystem and offer additional surface area for the attached organisms. In recent years, increasing attention has been risen on the profound effect on epiphytic bacterial communities (Gordon-Bradley et al. 2014; He et al. 2014; Hempel et al. 2008) and bacterioplankton communities in the water column in plant association (Zeng et al. 2012). Submerged macrophytes may directly impact the abundance and composition of planktonic bacterial communities via allelopathic effect or indirectly impact it via modulating the sedimentary resuspension and/or dissolved organic carbon and nitrogen cycling processes (Wu et al. 2007b). Field study and microcosm experiments showed that the variations of bacterioplankton community composition within lakes might be explained by the presence and maintenance of particular species of macrophytes (Zeng et al. 2012).
To epiphytic bacteria, immediate contact with leaves makes it more sensitive to the growth status of submerged macrophytes. Submerged macrophytes offer an additional niche for attached growth of microorganisms (Coci et al. 2008, 2010). On the other hand, submerged macrophytes release secondary metabolites during their life cycle and subsequently affect the construction and abundance of epiphytic bacteria (Hempel et al. 2008). The host plants, especially submerged macrophytes, have been estimated for their influence on the abundance and composition of epiphytic bacteria (Gordon-Bradley et al. 2014; He et al. 2012; Hempel et al. 2008, 2009) and bacterioplankton(Wu et al. 2007a; Zeng et al. 2012). The age of plant, leaf shape, allelochemically active polyphenols, etc. may cause variations in the diversity of bacterial community.
It has been pointed out that significant difference exists in the community structure between epiphytic bacteria and bacterioplankton even at the phylum level (He et al. 2012). However, another work reported that such difference in water column was only significant at species level rather than at community level (Gordon-Bradley et al. 2014). It is worth to mention that these works are mainly about epiphytic biofilms collected in single associations of submerged macrophytes, but the epiphytic bacterial community on different plant species from hybrid associations are seldom known.
Ceratophyllum demersum and Myriophyllum spicatum are highly allelophathically active, and species of Potamogeton genus are considered much less allelophathically active (Hilt and Gross 2008). Hemple et al. had revealed that the bacterial community structure and abundance had difference in the attached epiphyton between M. spicatum and Potamogeton perfoliatus (Hempel et al. 2009). However, no information is available concerning the difference between bacterial communities of epiphyton on C. demersum and Potamogeton malaianus.
Taihu Lake is a typical shallow lake with two states of alternative equilibria (Scheffer et al. 1993). Eastern Taihu Lake is dominated by submerged macrophytes. The diversity of epiphytic bacteria (He et al. 2012) and bacterioplankton (Zeng et al. 2012) and the geographical and seasonal change pattern of epiphytic bacteria attached on P. malaianus (Cai et al. 2013) have been reported. However, almost no studies comprehensively compared the seasonal change in patterns of epiphytic bacteria and bacterioplankton, especially nitrogen cycle bacteria such as denitrifying bacteria, in epiphytic and pelagic compartment of shallow fresh lake. Here, we conducted a comparative study of bacterial community in epiphyton of submerged plant P. malaianus and C. demersum as well as in bacterioplankton.
In this study, samples of epiphyton attached on two submerged macrophytes were collected in the same site in Taihu at four seasons, and bacterioplankton was simultaneously collected. Real-time quantitative PCR (qPCR) was applied to assess the copy numbers of nirK and nirS genes. Bacterial community structure was determined using Illumina MiSeq high-throughput sequencing. The aims of study were: (i) in the same background value (hybrid association) to estimate differences in the diversity patterns, including alpha and beta diversity, and bacterial community composition of in different compartments, (ii) to determine the abundance of nirK and nirS gene-bearing denitrifiers and assess the relationship between macrophytes species and denitrifiers, and (iii) to investigate whether host specificity or seasonal parameters affect the abundance and diversity of epiphytic bacteria.
Materials and methods
Study area
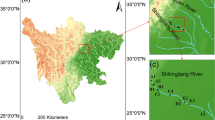
The sample site in this study was Taihu Lake, located in Jiangsu province, China. Being the third largest freshwater lake in China, the surface area of Taihu Lake is 2338 km2 with a mean depth of 1.89 m. Taihu Lake can be separated into two distinct ecological regions: the macrophyte-dominated zone and the phytoplankton-dominated zone. The sampling site of this research is allocated in the macrophyte-dominated lake zone. A large-scale aquatic macrophytes association located between Dongshan and Xishan islands in Taihu Lake (31° 5′ 9.21″ N, 120° 19′ 8.83″ E) were selected, which is composed by highly dense P. malaianus and C. demersum. P. malaianus and C. demersum are perennial submerged macrophytes, commonly seen from east to south Taihu Lake. Both macrophytes go into maturation period from June to October. The leaves of P. malaianus are long-shaped blade while that of C. demersum are wire-like with spiny teeth.
Sample collection and preparation
Submerged macrophytes and water were sampled in August, October of 2013 and January, April of 2014 which corresponded to florescence, young seedling, mature seedling, and decline stages of P. malaianus and C. demersum. All plant samples were cut from 15 to 30 cm of the canopy with a sickle. Each macrophyte species were collected with triplicates. Water samples were collected simultaneously within the association of each site at 15–30 cm below the water surface for bacterioplankton DNA extraction and water chemistry analysis. All samples were kept in icebox and shipped to laboratory within 6 h.
Water temperature (T), pH, and dissolved oxygen (DO) were measured in situ with HICHA HQ30D water quality monitor (HACH, USA.). Total nitrogen (TN) and total phosphorus (TP) were measured by hand follow national standard (HJ/T 199-2005 and GB 11893-1989), nitrate nitrogen (NO3 −–N), nitrite nitrogen (NO2 −–N), and ammoniacal nitrogen (NH4 +–N) were measured with AAC3 continuous flow analyzer (AutoAnalyzer 3, SEAL, German). All chemical analysis and DNA extraction were performed within 24 h.
DNA extraction
Healthy mature leaves (10 g) in same length were chosen for separating epiphyton. Epiphytic bacteria attached on macrophyte leaves were collected using the method previously described by Hemple et al. Epiphyton collection followed the approaches by Hemple et al. and He et al. Healthy mature leaves were obtained on a super clean bench with abacterial scissors and tweezers. Then put the plant tissue (2 g) in an abacterial 50-mL centrifuge tube with 40 mL cleaning fluid (2 mM PBS, 0.01 % v/v Tween 80) which was also disinfected. Five replicates were conducted for each sample. Put all centrifuge tubes into ultrasonic cleaner treated for 5 min, then shook at 120 rpm for 30 min in a shaking bed followed by ultrasonic oscillation for 5 min. Extracted plant tissue was then quickly removed with abacterial tweezers. The cleaning fluid was filtrated to 0.22-μm polycarbonate filters and kept at −20 °C. The DNA of epiphytic bacteria attached on filters was extracted with E.N.Z.A water DNA kit (OMEGA Bio-tek, Inc.) following the manual. Put the filter into a tube contains beads and buffer to homogenize and lyse the sample. After a heat-frozen step, humic acid, proteins, polysaccharides, and other contaminants subsequently precipitated. Add HTR reagent to remove those contaminants. Bind DNA in an HiBind® DNA spin-column and remove trace contaminants with two rapid wash steps. The pure DNA is eluted in low ionic strength buffer. The purified DNA can be directly used in downstream application without further purification. Bacterioplankton were collected from 1 L water samples with a two-time filtration: first 1.2 μm polycarbonate filters to remove particulate matter and phytoplankton then 0.22 μm polycarbonate filters to obtain bacterioplankton. The 0.22-μm polycarbonate filters were also kept at −20 °C until used. Total DNA of bacterioplankton was also extracted with E.N.Z.A water DNA kit (OMEGA Bio-tek, Inc.) followed manual. The size of extracted DNA was checked with 1 % agarose gels and quantified with Nanodrop 2000 (Thermo Scientific). The extracted DNA was stored at −20 °C for further analysis.
Quantitative PCR (qPCR)
nirK and nirS genes were used as molecular markers to determine the abundance of denitrifying bacteria. qPCR was performed in Step-one Plus Real-Time PCR Machine (Applied Biosystems) using SYBR Green method with two primers pair for nirK (F1aCu: 5′-ATCATGGTSCTGCCGCG-3′ / R3Gu: 5′- GCCTCGATCAGRTTGTGGTT-3′) and nirS (cd3a: 5′-GTSAACGTSAAGGARACSGG-3′ / R3cd: 5′-GASTTCGGRTGSGTCTTGA-3′) (Throback et al. 2004). Each 25 μL qPCR mixture contained 12.5 μL of TransStart® Top Green qPCR SuperMix (TransGen Biotech), 0.5 μL of each primer (10 μM), 0.5 μL of passive reference Dye (50×), and 10 ng of template DNA. The reactions were performed as 95 °C for 3 min and followed by 6 cycles of 95 °C for 30 s, 60 °C (nirK) or 57 °C (nirS) for 30 s, decrease 1 °C each cycles, 72 °C for 30 s (nirK) or 45 s(nirS), and 35 cycles of 95 °C for 30 s, 55 °C (nirK) or 52 °C (nirS) for 30 s, 72 °C for 30 s or 45 s (nirS), 4 °C forever at the end.
To determine gene abundances in one ng of extracted DNA, Hyphomicrobium denitrificans (nirK) and Pseudomonas fluorescens (nirS) strains were used to generate qPCR standard curves. All qPCR reactions including the standards were analyzed in triplicate. Before qPCR, a general PCR reaction was performed and the products were checked with 1 % agarose electrophoresis to confirm the specificity of primer pairs and qPCR products, and melt curves were also performed in each qPCR reaction. qPCR efficiency ranged from 80.1 to 107.6 % with R 2 values over 0.991 for all calibration curves. The abundance of denitrification genes = [sequence number of genes / total number of sequences/ DNA used in applications (ng)]*total DNA mass (ng) / plant or water sample weight (g).
Library construction and Illumina MiSeq high-throughput sequencing
Illumina MiSeq high-throughput sequencing was performed in Beijing Center for Physical and Chemical Analysis Co. Ltd (Beijing, China). PCR amplifications of bacterial V3 16S rRNA regions were carried out using the Phusion High-Fidelity PCR Master Mix (NewEast Biosciences, Inc., USA) and the primer pair 341F (5′-CCTACGGGAGGCAGCAG-3′) and 517R (5′-ATTACCGCGGCTGCTGG-3′). PCR amplification was carried out in triplicate using ABI 7500 Fast Real-Time PCR System in a total volume of 50 μl PCR reaction mix containing 5 μL 10× ExTaq Buffer, 4 μL dNTPs (2.5 mM), 0.25 μL ExTaq polymerase (TaKaRa Ex Taq), 10 pM of each primer, and 25 ng template DNA. Thermal cycling was performed at 94 °C for 5 min, 25 cycles at 94 °C for 1 min, 48 °C for 1 min, 72 °C for 1 min, and a final extension at 72 °C for 10 min. All the PCR products were performed with gel electrophoresis and purification. After purification, the amplicons were quantified with KAPA Library Quantification Kit. The purified amplicons were sequenced on an Illumina MiSeq platform (Illumina company). With FLASH, the paired-end reads were joined with an overlap length of 10 to 100 bps. The primer pairs were merged, and low quality sequences were also removed using U-Chime before Chimera detection. Sequences were assigned to the operational taxonomic units (OTUs) with a maximum distance of 3 %. Taxonomic assignment was performed using the RDP classifier (http://rdp.cme.msu.edu).
Statistical analysis
Community diversity indices (Chao1 estimator, Shannon index, and Simpson) and rarefaction curve of each sample were generated using the UPARSE pipeline. The difference in composition of species of three kinds of samples was compared with Venn (http://bioinformatics.psb.ugent.be/webtools/Venn/). PCoA (principal coordinates analysis) performed using R project for statistical computation was used to describe the relationship between different species or samples. The trend of the biological information matrix was calculated using Canoco v4.5 with detrended correspondence analysis (DCA), if the maximum length of four gradients in DCA result is above 4, CA, CCA, and DCA (unimodal) could be used in analysis, but if the length is less than 3, PCA and RDA (linear model) are more suitable, and if the length is between 3 and 4, both unimodal or linear model can be used (Leps and Smilauer 2003). In our study, the length of first gradient were 1.383 (P. malaianus), 1.205(C. demersum), and 1.302 (bacterioplankton), so redundancy analysis (RDA) was chosen to construct the relationship between species and environment factors.
Analysis of variance (one-way ANOVA) was used to determine the significance of difference (P < 0.05) using the software SPSS 20 in the abundance of functional genes among samples of each season. Pearson correlation coefficients and Spearman’s rank correlation coefficient (SRCC) were also performed to describe the relationship between denitrifying bacteria and environment factors.
Nucleotide sequence accession numbers
Sequences obtained in Illumina Miseq high-throughput sequencing were deposited in GenBank under accession nos SAMN04009790 to SAMN04009801, in the biosample no. SUB1073018.
Results
Environmental factors and nutrient conditions
The mean water temperature in sampling cites at four seasons from 2013 to 2014 were 18.35 °C (April), 29.4 °C (August), 18.6 °C (October), and 2.3 °C (January). pH and DO varied among sampling seasons (8.42∼8.57 and 7.12∼11.62 mg/L). Differences in nutrient were significant among seasons, and a slight increase of TN and TP was observed in April and August, though the trend of NO3 −, NO2 −, and NH4 + were unclear (Table 1).
Abundance of nirK and nirS genes
For the nirK gene in bacterioplankton communities, average abundances ranged from 6.98 × 106 to 2.40 × 108 copies/g water sample, whereas in epiphytic bacterial communities, the abundances were 1.55 × 104 to 3.23 × 1010copies/g (Dry weight [dw]) in epiphytic bacteria of P. malaianus and 9.83 × 103 to 8.95 × 1010copies/g dw of C. demersum. The abundance of nirS in bacterioplankton was 1.12 × 106 to 1.17 × 106 copies/g water sample, and the abundances in epiphytic bacteria were 1.68 × 106 to 2.38 × 107copies/g dw in P. malaianus and 1.30 × 107 to 2.89 × 107 copies/g dw in C. demersum.
The abundances of denitrification displayed remarkable distinction in different compartments. nirS gene abundance was higher in epiphytic bacteria attached on C. demersum than that on P. malaianus or in bacterioplankton (P < 0.05), but the opposite trend was observed in nirK gene (epiphyton of P. malaianus > C. demersum > bacterioplankton, P < 0.05) except in April where nirK gene abundance in epiphyton of C. demersum was higher than P. malaianus and bacterioplankton. Pearson correlation coefficients and SRCC revealed significantly negative correlation between nirS and nirK abundances were in epiphyton of C. demersum, but less significant in epiphyton of P. malaianus. However, positive correlation between the two groups of denitrifying bacteria was found in bacterioplankton communities (Table 2).
Remarkable seasonal variations in the abundance of nirK and nirS genes were also observed between epiphyton and bacterioplankton (Fig. 1). In the biofilm of P. malaianus and C. demersum, nirK gene abundance displayed a significant difference between April and the other three months (p < 0.01) where the folds of abundances were over 106 to 107. However, in bacterioplankton, the folds were only about 102. The seasonal changes of nirS gene were less conspicuous. In epiphyton, the abundance of nirS was higher in summer and winter and lower in spring and autumn, yet the folds were all less than 10. The seasonal variation in C. demersum was more inconspicuous than P. malaianus. The maximal fold in epiphyton of C. demersum between seasons (April/January) is only 2.27, which is 14.1 in the epiphyton of P. malaianus (October/January). The abundance of nirS in bacterioplankton was higher in spring and summer, but the lowest in winter; the maximal fold between seasons was 151 (January/April).

Quantitative analysis of nir gene in epiphyton and bacterioplankton samples. Error bars represent standard deviation calculated from three independent experiments. Difference between seasons based on ANOVA analysis shown in characters above error bars. (a Distribution of nirK gene in epiphyton attached on Potamogeton malaianus, Ceratophyllum demersum, and bacterioplankton; b distribution of nirS gene). Different indices indicate that there was a significant difference according to the Students–Newman–Keuls test with a confidence limit of 95 %
Effects of submerged macrophytes on denitrifying bacteria diversity and community composition
The microbial diversity and community structure of epiphyton and bacterioplankton were investigated with illumina MiSeq high-throughput sequencing. After removing low-quality sequences and chimeras, at least 58,181 effective sequences with average length of 151 bp were obtained in each sample. After normalized, using CD-HIT clustering method, each sample generated 1441–19,981 OTUs with the coverage is 0.83–0.98. However, the rarefaction curves did not reach the plateau, suggesting that there were still some species remained unidentified. All OTUs were assigned to 49 phyla and 485 genera. The number of OTUs assigned to phyla and genera were shown in Table 3. The unassigned reads were about 5.69–11.32 % of all OTUs (Table 3). For each community, the Shannon indexes ranged from 8.22 to 9.97, Simpson indexes ranged from 0.98 to 0.99, and Chao1 values from 3692.20 to 21652.93 (Table 4). Results of PCoA indicated that the bacterial communities were clustered into three groups according to sampling source but not seasons (Fig. 2).

Principal coordinates analysis (PCoA) for different samples based on all OTUs obtained in high-through sequencing
The major phyla in bacteria community were shown in Fig. 3a. The dominant phylum was Proteobacteria, which occupied 24.76 % (epiphyton of C. demersum in April) to 69.87 % (epiphyton of P. malaianus in August) of all OTUs. Alpha-, beta-, and gamma-proteobacteria were the dominant class of Proteobacteria occupied above 90 % of Proteobacteria. The following major phyla were Actinobacteria (average 22.31 %, 11.19– 47.94 % in each sample), Bacteroidetes (8.60 %, 3.91–16.90 %), Cyanobacteria (12.03 %, 5.04–37.80 %), and Firmicutes (1.52 %, 1.08–4.84 %). Although the abundances of phyla were significant different between host plants, the composition was similar. According to Venn diagram based on phyla and genera composition, only a few genera were distinctive between epiphyton and bacterioplankton (Fig. 4). About 67.3 % of all phyla were shared between different sources of microorganism samples. Three phyla were specific in epiphyton on P. malaianus and only 1 phylum was in bacterioplankton. However, 7 phyla were unique in the epiphyton from C. demersum which accounts 14.3 % of the total number of phyla (Fig. 4a). However, compare with phyla, much less proportion of genera are overlapped in both bacterioplankton and epiphyton: total 288 genera shared between different samples, about 59.4 % of total communities (Fig. 4b). More genera overlapped between two kinds of samples, as 8.2 % genera shared between bacterioplankton and epiphyton of P. malaianus, 7.5 % shared between bacterioplankton and epiphyton of C. demersum, and 6 % shared between two kinds of epiphyton, respectively. Less particular genera were found in epiphyton of P. malaianus and bacterioplankton (5, accounts 1 % of total genera) and bacterioplankton (3, 0.6 % of total genera), but more in epiphyton of C. demersum (85, 17.5 % of total genera).

Distribution of bacterial in different samples at phylum level (a), percentages of the major microorganism based on genera in each sample (b), and distribution of core genera of denitrifying bacteria (c). The relative abundances of these bacteria were above 3 % of whole community. P, epiphyton on Potamogeton malaianus; C, Ceratophyllum demersum; B, bacterioplankton

Venn diagram base on phyla (a) and genera (b) in different samples. Three circles represent epiphyton on Potamogeton malaianus, Ceratophyllum demersum, and bacterioplankton. The overlapping area is phyla shown in two to three samples. The number in different area is the number of phyla or genera
Identification of core denitrifying genera, nirK and nirS analysis
Significant differences in microorganisms’ abundance between different samples were found in accordance with the bacteria taxonomic distribution (Fig. 3). Above all, Pseudomonas and Flavobacterium were the major genera of bacterial community (Fig. 3b). As to denitrifying bacteria, Pseudomonas was the core denitrifying bacteria genus in epiphyton of P. malaianus and bacterioplankton, which occupied 29.29–34.85 % of total count of OTUs in the epiphytic compartment of P. malaianus and about 36.01–39.49 % of pelagic compartment (Fig. 3c). However, in the epiphyton of C. demersum, Pseudomonas was only 5.39–6.16 % of all sequence. Rhodobacter and Bacillus were the co-dominant denitrifying genera which occupied about 3.42–9.59 % and 4.82–8.23 % of all sequences obtained in epiphytic bacteria attached on C. demersum, although these two genera were only 2.06–4.33 % and 0–0.11 % of whole community in the epiphyton of P. malaianus and even less in pelagic compartment (1.48–2.67 % and 0.02–0.06 %). Besides, Micrococcus also showed unique distribution in different samples, which were 1.03–1.29 % in bacterial community of epiphyton of C. demersum, but only 0.04–0.11 % of epiphytic compartment of P. malaianus and 0.06–0.17 % of pelagic compartment.
Relationship between environment factors and denitrifying bacteria community structure
Based on the SRCC, the relationships between environment factors and the abundance of nirK and nirS were calculated (Table 2). nirK gene showed significantly positive correlation with TN(P < 0.05) and negative correlation with NH4 +–N (P < 0.05), NO2 −–N, and pH (P < 0.5). However, nirS may referred to different limiting factors in different samples: the abundances of nirS gene in epiphyton positively correlated with NH4 +–N and pH (P < 0.05); at the same time, it also positively correlated with NO2 —N (P < 0.05) in epiphyton samples of C. demersum. However, in bacterioplankton samples, nirS gene positively correlated with TN and TP (P < 0.5) and negatively with NH4 +–N (P < 0.5). Although there are differences between results of Pearson correlation coefficients and SRCC, the general trend of correlation is conformable.
Due to the lengths of first gradient were less than 3 (1.56 in P. malaianus, 1.68 in C. demersum, and 1.46 in bacterioplankton) in DCA, RDA, a kind of constrained ordination was chosen to analyze the relationship between the relative abundance of denitrifying bacteria (genera level) and the environment factors. As displayed in Fig. 5, the first and second canonical axes represented 88.8 and 11.1 % of variation in P. malaianus, which were 98.9 and 0.7 % in C. demersum and 98.4 and 1.5 % in bacterioplankton respectively. Same genus was affected by different environment factors in epiphyton and bacterioplankton. The four dominant genera: Pseudomonas, Rhodobacter, Bacillus, and Agrobacterium assembled in one cluster which related to pH and NH4 +–N in epiphyton of C. demersum, and Micrococcus was independent outside and related to NO3 −–N. Although the cluster structure in bacterioplankton was similar to the structure of epiphyton of C. demersum, the relationship between those denitrifying bacteria and environment factors was unclear. Unlike those the cluster structure, Micrococcus, Rhodobacterium, Bacillus, and Agrobacterium assemble in one cluster, and Pseudomonas was independent in the epiphytic bacteria on P. malaianus, but the relationship between them and environment was inconspicuous.

RDA to investigate the ecological correlation between abundance of nir gene, microorganism and environmental parameters. (a) Epiphyton of Potamogeton malaianus; (b) epiphyton of Ceratophyllum demersum; (c) bacterioplankton (round dot for microorganism based on genera; arrow for environmental parameters)
Discussion
Abundance of nirK and nirS gene
In freshwater ecosystems, there exists extensive denitrification activity in the epiphyton of submerged macrophytes (Eriksson 2001; Eriksson and Weisner 1997). High density of denitrifiers attached to submerged macrophytes was calculated with MPN in previous studies (Eriksson 2001; Körner 1999). In Wuhle River, which contains a treated sewage channel, the abundance of denitrifiers were 9.4 × 104 ± 7 × 104 MPN mL−1 in water and 3.3 × 106 ± 2.5 × 106 MPN mL−1 in sediment, but the averaged numbers of denitrifiers was higher (8.7 × 107 ± 1 × 107 MPN g−1 (plant DW)) in the epiphytic communities of six species of submerged macrophytes which included C. demersum, P. matans, and P. crispus (Körner 1999). These results suggested that the abundances of denitrifiers were higher in epiphyton than in benthic and pelagic compartments in eutrophic water body. In our study, abundance of nirS gene was higher in epiphytic bacterial than bacterioplankton, suggesting that the epiphyton may work as a niche for the denitrifying bacteria with nirS.
In eutrophic lake like Donghu Lake, the abundance of nirS varied from 1.47 × 104 to 1.67 × 105 copies/g dry sediment, and nirK abundance varied from 2.02 × 107 to 9.71 × 107 copies/g dry sediment (Hou et al. 2013). The quantity of nirS gene in epiphyton was lower than that in Wuhle River, and higher than in the sediment of Donghu Lake, but nirK gene abundances were lower than previous studies. In previous studies, the abundance of nirK was often higher than nirS in sediment (Hou et al. 2013; Saarenheimo et al. 2015), epiphytic biofilms (Vila-Costa et al. 2014), and soil (Barta et al. 2010; Dandie et al. 2011; Su et al. 2010). However, in our study except April, nirS was generally higher than nirK, similar to the trends reported in agricultural ecosystems (Attard et al. 2011; Petersen et al. 2012).
Submerged macrophytes have been reported to work as a niche to nitrogen cycling bacteria (Coci et al. 2010). nirK and nirS genes were detected on epiphyton which means favorable physicochemical conditions over macrophytes surface provide a niche for denitrifying bacteria. Worth to notice, nirK and nirS genes showed different distribution in epiphyton and bacterioplankton. nirK gene abundance was bacterioplankton > P. malaianus > C. demersum, while nirS gene showed a different trend: C. demersum > P. malaianus > bacterioplankton. Difference in leaf structure may also affect the attached bacterial community (Gordon-Bradley et al. 2014). C. demersum has small, feathery leaves, which provides more protective habitat for bacterial colonizers. The leaves of P. malaianus were flat, broad and straight, which could be a challenging habitat for some specific bacteria that did not prefer attached growth. Besides, different ability of producing secondary compounds like polyphenols and cyclic sulfur compounds may also affect epiphyton on Ceratophyllum and Potamogeton macrophytes (Hilt and Gross 2008). Isoenzyme encoding genes have different habitat preference is not unusual too. In marine environment, algal provided a specific ecological niche for ammonia-oxidizing bacteria (AOB), but to ammonia-oxidizing archaea (AOA), lower abundance suggested specific positive and negative epiphyte–host interactions (Trias et al. 2012). In our study, high abundance of nirS and low abundance of nirK suggest that nirK-type denitrifiers are more sensitive to environmental variation than nirS-type denitrifiers (Santoro et al. 2006; Smith and Ogram 2008; Throback et al. 2004; Wolsing and Prieme 2004; Yuan et al. 2012).
Difference in bacterial community composition
Generally, potential interactions may exist between submerged macrophytes and epiphytic microorganism, which is known as host specificity. Many reports described diversity of epiphytic bacteria on submerged macrophytes and of surrounding bacterioplankton. The perspective was well accepted that epiphytic bacteria exhibited higher diversity and distinct community composition than the surrounding bacterioplankton (He et al. 2014). The presence of bacteria is also dependent on macrophyte species (Hempel et al. 2008), such as in functional bacteria AOA and AOB (Coci et al. 2010; Trias et al. 2012). However, with Illumina MiSeq high-throughput sequencing, highly consistent community composition was found in two epiphytic bacteria and in bacterioplankton in our study. This result was similar to the find of Gordon-Bradley et al. (2014), attributing to all samples come from one mixed-submerged macrophytes community. However, we also found significant difference of bacterial abundance, especially some dominant phyla that were distinct among sample sources (epiphyton/bacterioplankton) and the species of macrophytes (P. malaianus/C. demersum).
The phylum Protebacteria dominated in epiphytic bacteria and occupied above 24.76 % of all sequences. It was also found to play a dominant role in epiphytic bacteria of other aquatic plants (Burke et al. 2011; Crump and Koch 2008; He et al. 2012, 2014; Hempel et al. 2008; Tujula et al. 2010). As to bacterioplankton, Actinobacteria was the main phylum and occupied above 38.99 % of all sequences, which is widely ranged in Taihu Lake (Zeng et al. 2012) and other freshwater bodies (Bruns et al. 2003; He et al. 2014; Jezberova et al. 2010; Kasalicky et al. 2013) but much less frequently discovered in epiphytic bacteria. The difference of epiphytic bacterial community and bacterioplankton can be concluded into different phylogenetic resolutions (He et al. 2014), which agreed with marine green alga (Burke et al. 2011). The epiphytic bacteria on the two kinds of submerged macrophytes in our study also had similar community composition, and epiphytic bacteria community still showed distinguishable host specificity in bacteria abundance. For example, Protebacteria has higher abundance in the epiphyton of P. malaianus than in C. demersum while Actinobacteria and Cyanobacteria were oppositely more abundant in C. demersum. Similar studies also revealed difference between the epiphytic bacterial communities on plants (Gordon-Bradley et al. 2014; He et al. 2012; Hempel et al. 2008, 2009).
Denitrifying bacteria also presented a similar composition, though host specificity can still be found in genera abundance. In epiphyton of P. malaianus and bacterioplankton, Pseudomonas dominates the most important part of denitrifying bacteria and second with Rhodobacter. However, in the epiphytic bacteria of C. demersum, the dominant positions of these two genera were not stable as Bacillus showed really high abundance. Besides, the three genera occupied 31.66–38.87 % of the whole bacterial community in epiphyton of P. malaianus and in bacterioplankton respectively, but was only 14.40–25.14 % in epiphytic bacterial community of C. demersum. In epiphyton of V. natans and H. verticillata, betaproteobacteria has the highest abundance among all bacteria (He et al. 2012). To combine the nirK- and nirS gene-bearing denitrifiers abundance abovementioned, species of submerged macrophytes (host specificity) and sample source may strongly influence the niche and composition of denitrifiers.
Relationship between denitrifiers and environmental parameters
The relationship between denitrifier abundance, community structure, and environmental properties or denitrification or N2O emissions has been investigated under a range of environment showing varied results (Dandie et al. 2011; Enwall et al. 2010; Philippot et al. 2009; Rich and Myrold 2004; Wertz et al. 2006). In previous studies in agricultural system, the opposite relationship was found that the abundance nirS gene-bearing denitrifiers was related to pH, NH4 +–N and NO3 −–N concentrations, while no such relationships between environmental properties and nirK gene-bearing denitrifiers abundance were observed (Philippot et al. 2009). nirS gene structure being significantly correlated with pH, but nirK gene community structure was not (Enwall et al. 2010). Pseudomonas mandelii and closely related spp. abundance showed low but significantly positive correlations with NO3 −–N of adjacent riparian and agricultural zones (Dandie et al. 2011). In our study, nirS- and nirK-bearing denitrifiers showed a correlation with TN, NO2 −–N, NH4 +–N, and pH in correlation analysis, but some different coefficient of correlation was also obtained. Bacterial community was explained better by host specificity than seasonal and environmental parameters for samples assembled according to sample source but not seasonal variation in PCoA (Fig. 2). In RDA, different relationships were obtained between main denitrifying genera and environmental variables (Fig. 5). In bacterioplankton and epiphytic bacteria of P. malaianus, no significant correlation between dominant denitrifying genera and environmental properties was observed, but significantly positive correlation was found between dominant denitrifying genera and environmental variables. Different coefficients of correlation of nirS- and nirK-bearing denitrifier and community structure may due to different ecological redundancy (Hou et al. 2013) and environmental sensitivity (Smith and Ogram 2008) and different processes define the niche that denitrifiers occupying and the factors controlling their diversity (Dandie et al. 2011). Since the driving mechanism between formation mechanism of niche and denitrifiers was still unclear, further study needs to emphasize on the role of nirK- and nirS gene-bearing denitrifiers in the denitrification process.
Conclusion
We simultaneously studied the nirK- and nirS gene-bearing denitrifiers in bacterioplankton and epiphyton on P. malaianus and C. demersum in Taihu Lake. nirK and nirS gene dominated the bacterioplankton and epiphyton samples, respectively. nirS gene showed higher gene copy numbers in epiphyton than in bacterioplankton, although nirK was lower, implicating difference of niche of these two denitrifying genes. In addition, although the bacterioplankton community composition was highly similar to epiphytic bacterial communities, the abundance of core phyla and genera was different among three sources. Furthermore, the main genera of denitrifiers also showed significantly difference between bacterioplankton and epiphyton attached on two submerged macrophytes, which means host specificity has a significant power in shaping bacterial/denitrifiers community. No significantly seasonal variation was found in bacterioplankton and epiphytic bacterial communities; however, environmental parameters played an important effect in nirK and nirS gene abundances. Further study is needed to investigate the relative contribution of nirK- and nirS gene-bearing denitrifiers to the denitrification process in bacterioplankton and epiphyton.
Abbreviations
- DO:
-
Dissolved oxygen
- TN:
-
Total nitrogen
- TP:
-
Total phosphorus
- NO3 −–N:
-
Nitrate nitrogen
- NO2 −–N:
-
Nitrite nitrogen
- NH4 +–N:
-
Ammoniacal nitrogen
- qPCR:
-
Quantitative PCR
- OTUs:
-
Operational taxonomic units
- PCoA:
-
Principal coordinates analysis
- DCA:
-
Detrended correspondence analysis
- RDA:
-
Redundancy analysis
- SRCC:
-
Spearman’s rank correlation coefficient
- DW:
-
Dry weight
- AOA:
-
Ammonia-oxidizing archaea
- AOB:
-
Ammonia-oxidizing bacteria
References
Abell GC, Revill AT, Smith C, Bissett AP, Volkman JK, Robert SS (2010) Archaeal ammonia oxidizers and nirS-type denitrifiers dominate sediment nitrifying and denitrifying populations in a subtropical macrotidal estuary. ISME Journal 4:286–300. doi:10.1038/ismej.2009.105
Antonyuk SV, Strange RW, Sawers G, Eady RR, Hasnain SS (2005) Atomic resolution structures of resting-state, substrate- and product-complexed Cu-nitrite reductase provide insight into catalytic mechanism. Proc Natl Acad Sci U S A 102:12041–12046
Attard E et al (2011) Soil environmental conditions rather than denitrifier abundance and diversity drive potential denitrification after changes in land uses. Glob Chang Biol 17:1975–1989
Barta J, Melichova T, Vanek D, Picek T, Santruckova H (2010) Effect of pH and dissolved organic matter on the abundance of nirK and nirS denitrifiers in spruce forest soil. Biogeochemistry 101:123–132
Bowen JL, Babbin AR, Kearns PJ, Ward BB (2014) Connecting the dots: linking nitrogen cycle gene expression to nitrogen fluxes in marine sediment mesocosms. Front Microbiol 5:429. doi:10.3389/fmicb.2014.00429
Braker G, Fesefeldt A, Witzel KP (1998) Development of PCR primer systems for amplification of nitrite reductase genes (nirK and nirS) to detect denitrifying bacteria in environmental samples. Appl Environ Microbiol 64:3769–3775
Braker G, Zhou J, Wu L, Devol AH, Tiedje JM (2000) Nitrite reductase genes (nirK and nirS) as functional markers to investigate diversity of denitrifying bacteria in pacific northwest marine sediment communities. Appl Environ Microbiol 66:2096–2104
Braker G, Schwarz J, Conrad R (2010) Influence of temperature on the composition and activity of denitrifying soil communities. FEMS Microbiol Ecol 73:134–148
Bremer C, Braker G, Matthies D, Reuter A, Engels C, Conrad R (2007) Impact of plant functional group, plant species, and sampling time on the composition of nirK-Type denitrifier communities in soil. Appl Environ Microbiol 73:6876–6884
Bruns A, Nubel U, Cypionka H, Overmann J (2003) Effect of signal compounds and incubation conditions on the culturability of freshwater bacterioplankton. Appl Environ Microbiol 69:1980–1989
Burke C, Thomas T, Lewis M, Steinberg P, Kjelleberg S (2011) Composition, uniqueness and variability of the epiphytic bacterial community of the green alga Ulva australis. ISME Journal 5:590–600. doi:10.1038/ismej.2010.164
Cai X, Gao G, Tang X, Dong B, Dai J, Chen D, Song Y (2013) The response of epiphytic microbes to habitat and growth status of Potamogeton malaianus Miq. in Lake Taihu. J Basic Microbiol 53:828–837
Coci M, Bodelier PLE, Laanbroek HJ (2008) Epiphyton as a niche for ammonia-oxidizing bacteria: detailed comparison with benthic and pelagic compartments in shallow freshwater lakes. Appl Environ Microbiol 74:1963–1971
Coci M, Nicol GW, Pilloni GN, Schmid M, Kamst-van Agterveld MP, Bodelier PLE, Laanbroek HJ (2010) Quantitative assessment of ammonia-oxidizing bacterial communities in the epiphyton of submerged macrophytes in Shallow Lakes. Appl Environ Microbiol 76:1813–1821
Crump BC, Koch EW (2008) Attached bacterial populations shared by four species of aquatic angiosperms. Appl Environ Microbiol 74:5948–5957. doi:10.1128/AEM.00952-08
Dandie CE et al (2011) Abundance, diversity and functional gene expression of denitrifier communities in adjacent riparian and agricultural zones. FEMS Microbiol Ecol 77:69–82
Enwall K, Philippot L, Hallin S (2005) Activity and composition of the denitrifying bacterial community respond differently to long-term fertilization. Appl Environ Microbiol 71:8335–8343. doi:10.1128/AEM.71.12.8335-8343.2005
Enwall K, Throback IN, Stenberg M, Soderstrom M, Hallin S (2010) Soil resources influence spatial patterns of denitrifying communities at scales compatible with land management. Appl Environ Microbiol 76:2243–2250
Eriksson PG (2001) Interaction effects of flow velocity and oxygen metabolism on nitrification and denitrification in biofilms on submersed macrophytes. Biogeochemistry 55:29–44. doi:10.1023/a:1010679306361
Eriksson PG, Weisner SEB (1997) Nitrogen removal in a wastewater reservoir: the importance of denitrification by epiphytic biofilms on submersed vegetation. J Environ Qual 26:905–910
GB11893-1989: Water quality- determination of total phosphorus- ammonium molybdate spectrophotometric method. http://www.mep.gov.cn/image20010518/3655.pdf
Gordon-Bradley N, Lymperopoulou DS, Williams HN (2014) Differences in bacterial community structure on Hydrilla verticillata and Vallisneria americana in a freshwater spring. Microbes Environments JSME 29:67–73
Hai B et al (2009) Quantification of key genes steering the microbial nitrogen cycle in the rhizosphere of sorghum cultivars in tropical agroecosystems. Appl Environ Microbiol 75:4993–5000. doi:10.1128/AEM.02917-08
He D, Ren LJ, Wu QL (2012) Epiphytic bacterial communities on two common submerged macrophytes in Taihu Lake: diversity and host-specificity. Chin J Oceanol Limnol 30:237–247. doi:10.1007/s00343-012-1084-0
He D, Ren LJ, Wu QLL (2014) Contrasting diversity of epibiotic bacteria and surrounding bacterioplankton of a common submerged macrophyte, Potamogeton crispus, in freshwater lakes. FEMS Microbiol Ecol 90:551–562
Hempel M, Blume M, Blindow I, Gross EM (2008) Epiphytic bacterial community composition on two common submerged macrophytes in brackish water and freshwater. BMC Microbiol 8:58. doi:10.1186/1471-2180-8-58
Hempel M, Grossart HP, Gross EM (2009) Community composition of bacterial biofilms on two submerged macrophytes and an artificial substrate in a pre-alpine lake. Aquat Microb Ecol 58:79–94
Hilt S, Gross EM (2008) Can allelopathically active submerged macrophytes stabilise clear-water states in shallow lakes? Basic Appl Ecol 9:422–432
HJ/T199-2005: Water quality- determination of total-nitrogen gas-phase molecular absorption spectrometry. http://kjs.mep.gov.cn/hjbhbz/bzwb/shjbh/sjcgfffbz/200601/W020110127355110870522.pdf
Hou J, Cao X, Song C, Zhou Y (2013) Predominance of ammonia-oxidizing archaea and nirK-gene-bearing denitrifiers among ammonia-oxidizing and denitrifying populations in sediments of a large urban eutrophic lake (Lake Donghu). Can J Microbiol 59:456–464. doi:10.1139/cjm-2013-0083
Jezberova J, Jezbera J, Brandt U, Lindstrom ES, Langenheder S, Hahn MW (2010) Ubiquity of Polynucleobacter necessarius ssp. asymbioticus in lentic freshwater habitats of a heterogeneous 2000 km area. Environ Microbiol 12:658–669. doi:10.1111/j.1462-2920.2009.02106.x
Kasalicky V, Jezbera J, Hahn MW, Simek K (2013) The diversity of the Limnohabitans genus, an important group of freshwater bacterioplankton, by characterization of 35 isolated strains. PLoS One 8:e58209. doi:10.1371/journal.pone.0058209
Körner S (1999) Nitrifying and denitrifying bacteria in epiphytic communities of submerged macrophytes in a treated sewage channel. Acta Hydrochim Hydrobiol 27:27–31
Kraft B, Tegetmeyer HE, Meier D, Geelhoed JS, Strous M (2014) Rapid succession of uncultured marine bacterial and archaeal populations in a denitrifying continuous culture. Environ Microbiol 16:3275–3286. doi:10.1111/1462-2920.12552
Leps J, Smilauer P (2003) Multivariate analysis of ecological data using CANOCO. United States of America by Cambridge University Press, New York
Petersen DG, Blazewicz SJ, Firestone M, Herman DJ, Turetsky M, Waldrop M (2012) Abundance of microbial genes associated with nitrogen cycling as indices of biogeochemical process rates across a vegetation gradient in Alaska. Environ Microbiol 14:993–1008
Philippot L et al (2009) Mapping field-scale spatial patterns of size and activity of the denitrifier community. Environ Microbiol 11:1518–1526
Rich JJ, Myrold DD (2004) Community composition and activities of denitrifying bacteria from adjacent agricultural soil, riparian soil, and creek sediment in Oregon, USA. Soil Biol Biochem 36:1431–1441
Saarenheimo J, Rissanen AJ, Arvola L, Nykanen H, Lehmann MF, Tiirola M (2015) Genetic and environmental controls on nitrous oxide accumulation in lakes. PLoS One 10:e0121201. doi:10.1371/journal.pone.0121201
Santoro AE, Boehm AB, Francis CA (2006) Denitrifier community composition along a nitrate and salinity gradient in a coastal aquifer. Appl Environ Microbiol 72:2102–2109. doi:10.1128/AEM.72.3.2102-2109.2006
Scheffer M, Hosper SH, Meijer ML, Moss B, Jeppesen E (1993) Alternative equilibria in shallow lakes. Trends Ecol Evol 8:275–279. doi:10.1016/0169-5347(93)90254-M
Smith JM, Ogram A (2008) Genetic and functional variation in denitrifier populations along a short-term restoration chronosequence. Appl Environ Microbiol 74:5615–5620. doi:10.1128/AEM.00349-08
Su MX, Kleineidam K, Schloter M (2010) Influence of different litter quality on the abundance of genes involved in nitrification and denitrification after freezing and thawing of an arable soil. Biol Fertil Soils 46:537–541
Szukics U, Abell GCJ, Hodl V, Mitter B, Sessitsch A, Hackl E, Zechmeister-Boltenstern S (2010) Nitrifiers and denitrifiers respond rapidly to changed moisture and increasing temperature in a pristine forest soil. FEMS Microbiol Ecol 72:395–406
Throback IN, Enwall K, Jarvis A, Hallin S (2004) Reassessing PCR primers targeting nirS, nirK and nosZ genes for community surveys of denitrifying bacteria with DGGE. FEMS Microbiol Ecol 49:401–417. doi:10.1016/j.femsec.2004.04.011
Trias R, Garcia-Lledo A, Sanchez N, Lopez-Jurado JL, Hallin S, Baneras L (2012) Abundance and composition of epiphytic bacterial and archaeal ammonia oxidizers of marine red and brown macroalgae. Appl Environ Microbiol 78:318–325. doi:10.1128/aem.05904-11
Tujula NA, Crocetti GR, Burke C, Thomas T, Holmstrom C, Kjelleberg S (2010) Variability and abundance of the epiphytic bacterial community associated with a green marine Ulvacean alga. ISME Journal 4:301–311. doi:10.1038/ismej.2009.107
Vila-Costa M, Bartrons M, Catalan J, Casamayor EO (2014) Nitrogen-cycling genes in epilithic biofilms of oligotrophic high-altitude lakes (central Pyrenees, Spain). Microb Ecol 68:60–69. doi:10.1007/s00248-014-0417-2
Wang Z, Zhang XX, Lu X, Liu B, Li Y, Long C, Li A (2014) Abundance and diversity of bacterial nitrifiers and denitrifiers and their functional genes in tannery wastewater treatment plants revealed by high-throughput sequencing. PLoS One 9:e113603. doi:10.1371/journal.pone.0113603
Wertz S et al (2006) Maintenance of soil functioning following erosion of microbial diversity. Environ Microbiol 8:2162–2169
Wolsing M, Prieme A (2004) Observation of high seasonal variation in community structure of denitrifying bacteria in arable soil receiving artificial fertilizer and cattle manure by determining T-RFLP of nir gene fragments. FEMS Microbiol Ecol 48:261–271. doi:10.1016/j.femsec.2004.02.002
Wu QL, Zwart G, Wu J, Agterveld MPKV, Liu S, Hahn MW (2007a) Submersed macrophytes play a key role in structuring bacterioplankton community composition in the large, shallow, subtropical Taihu Lake, China. Environ Microbiol 9:2765–2774
Wu QL, Zwart G, Wu J, Kamst-van Agterveld MP, Liu S, Hahn MW (2007b) Submersed macrophytes play a key role in structuring bacterioplankton community composition in the large, shallow, subtropical Taihu Lake. China Environ Microbiol 9:2765–2774. doi:10.1111/j.1462-2920.2007.01388.x
You SJ (2005) Identification of denitrifying bacteria diversity in an activated sludge system by using nitrite reductase genes. Biotechnol Lett 27:1477–1482
Yuan Q, Liu P, Lu Y (2012) Differential responses of nirK- and nirS-carrying bacteria to denitrifying conditions in the anoxic rice field soil. Environ Microbiol Rep 4:113–122. doi:10.1111/j.1758-2229.2011.00311.x
Zeng J, Bian YQ, Xing P, Wu QLL (2012) Macrophyte species drive the variation of bacterioplankton community composition in a shallow freshwater lake. Appl Environ Microbiol 78:177–184
Zhang JX, Yang YY, Zhao L, Li YZ, Xie SG, Liu Y (2015) Distribution of sediment bacterial and archaeal communities in plateau freshwater lakes. Appl Microbiol Biotechnol 99:3291–3302
Zumft WG (1997) Cell biology and molecular basis of denitrification. Microbiol Mol Biol Rev 61:533–616
Acknowledgments
The authors are grateful to Ms. Ya-peng Zhang and Mr. Xun Cao for their valuable help in field investigation and sampling. This work is financially supported by the National Natural Science Foundation of China (41173078 & 41403064), the Natural Science Foundation of Jiangsu Province, China (BK20140922), and by the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (14KJB610007).
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Robert Duran
Rights and permissions
About this article

Cite this article
Fan, Z., Han, Rm., Ma, J. et al. Submerged macrophytes shape the abundance and diversity of bacterial denitrifiers in bacterioplankton and epiphyton in the Shallow Fresh Lake Taihu, China. Environ Sci Pollut Res 23, 14102–14114 (2016). https://doi.org/10.1007/s11356-016-6390-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-016-6390-1