
Abstract
This study is the first to investigate the simultaneous presence of NH4 + and fluorescent organic matter components (FOCs) from a fluvio-lacustrine aquifer in Central Jianghan Plain. Sediment, groundwater, and surface water samples were collected for the sediment organic matter extraction, 3D fluorescence spectroscopy characterization, and/or hydrochemical analysis. NH4 + and dissolved organic carbon was ubiquitous in the groundwater. The fluorescence spectroscopy revealed good relationships between NH4 + and fulvic acid-like components (FALCs) in the groundwater and sediment-extracted organic matter (SEOM) solutions. NH4 + also exhibited significant positive correlation with protein-like component (PLC) (p < 0.001), with the stronger in the SEOM solutions than that in groundwater. Comparisons of spectroscopic indices [e.g., humification index (HIX), biological index (BIX), spectra slope (S275–295), and specific UV absorbance (SUVA254)] between the groundwater and SEOM solutions revealed more labile properties of SEOM. This result indicates that the decreasing NH4 +–FOCs correlations of groundwater relative to sediments may be attributed to microbial degradation. Factor analysis identifies important factors that cause NH4 + occurrence in the groundwater. The accompanying increase of FALC (C1) and NH4–N with the mole concentration of the normalized HCO3 −/(Ca2++Mg2+) and [H+] suggests that couple effects of various biodegradations simultaneously occur in the aquifer, promoting the occurrence of NH4–DOMs.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Nitrogen speciation in the environment plays key roles in maintaining life activity and regulating the nitrogen balance in the ecosystem. NO3 − and NH4 + are the two most important forms of reactive nitrogen and the main nitrogen species of concern. Groundwater NO3 − has been extensively investigated worldwide (Hinkle and Tesoriero 2014; Spalding and Exner 1993); however, groundwater NH4 + has received relatively less research attention. Despite having no direct risk to human health, a high concentration of NH4 + can potentially increase NO3 − concentration via nitrification (Umezawa et al. 2008), which in turn leads to eutrophication of recharged surface water. In special conditions, groundwater NH4 + can indicate the presence of hazardous contaminants, such as pathogens and pesticides (Umezawa et al. 2008). Therefore, ammonium is regulated as a contaminant in several countries, including the UK and China; both countries prescribe a 0.5 mg/L maximum limit concentration of ammonium in drinking water (PRC Ministry of Health and Standardization Administration 2006; Erskine 2000).
NH4 + indicates the reducing condition of groundwater, and its occurrence has been commonly identified to be attributed to the denitrification of infiltrated NO3 − (anthropogenic source) (Stumm and Morgan 1996). Recently, the natural occurrence of NH4–N in aquifers, which may be easily overlooked, has been increasingly documented. Jiao et al. (2010) found abnormally high levels of NH4 + in the coastal aquifer system of the Pearl River Delta. Their study suggests that NH4 + is related to the high content of organic nitrogen in organic matter (OM)-rich aquitards. NH4 + is also identified as one of the main nutrients that are released from sedimentary organic matter (SOM) through submarine groundwater discharge processes (Gleeson et al. 2013; Santos et al. 2008). Most of the relevant observations are from coastal aquifers (Manning and Hutcheon 2004). Few studies have suggested the natural occurrence of ammonium in fresh groundwater (Glessner and Roy 2009; Hinkle and Tesoriero 2014). However, the detailed OM source information has not been specifically examined, especially the associations of NH4 + with OM components between sediments and groundwater.
Jianhan Plain is a large-scale river basin in Central China that is formed by the Yangtze and Han rivers (Duan et al. 2015; Huang et al. 2013). Widespread lacustrine sediments with abundant OM are observed in the low-flat part of the plain in the middle reaches of the Yangtze River (Huang et al. 2013). Previous studies showed the reducing quality of groundwater in the study area as evidenced by the reductive hydrochemical indices. Significant levels of NH4 +, bicarbonate, and dissolved organic carbon (DOC) were ubiquitous in the groundwater. Given the relatively closed nature of the aquifer and the prevalence of NH4 + and DOC, we inferred that observed anthropogenic point sources (typically shallow or surface NO3 −) unlikely cause the widespread NH4 + in groundwater. Therefore, we hypothesize that the occurrence of NH4 + may be linked to the OM of aquifers.
Given that the presence of NH4 + in groundwater may be related to OM source, the relationships between NH4 + and specific OM components should be determined. 3D fluorescence spectroscopy is a simple and convenient method that can provide a considerable amount of information on the composition and structure of DOM (Hur and Kim 2009; Wang et al. 2014). This method is highly sensitive at natural abundance levels and has been widely used to assess the quality of DOM in various aquatic systems (Fellman et al. 2010). By using this method, we report the concurrence of NH4 + and fluorescent OM components (FOCs) in the groundwater of a fluvio-lacustrine aquifer in Jianghan Plain, where widespread occurrence of DOC was believed to influence its groundwater chemistry significantly (Huang et al. 2015; Huang et al. 2012). The presented relationships of specific FOC with NH4 + and characteristic hydrochemical parameters provide new insights into the NH4 + genesis in an OM-rich lacustrine aquifer. Our approach for determining the occurrence of NH4 + in groundwater may be applicable to other regions that have similar conditions.
Materials and methods
Study area and sample collection
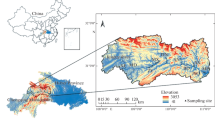
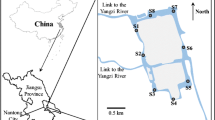
The study was conducted in the town of ShaHu in the Central Jianhan Plain (30° 08′ N, 113° 40′ E) (Fig. 1) where the reducing condition of groundwater driven by organic carbon was extensively documented (Deng et al. 2014; Huang et al. 2012). The sample collection was conducted within an interstream area that was dominated by lacustrine facies and currently developed as farmlands and fishery ponds. The shallow aquifers comprise gray sandy silts of 7.2 to 12 m and the gray fine and yellowish medium sands below 17.2 m (Fig. 1). The textures of the upper overlaying lacustrine clayey materials led to the reducing condition of the groundwater in the area. The Dongjing River, Tongshun River, and several streams meander through the area. The groundwater tables exhibit seasonal fluctuations with around 2 months delay relative to the precipitation cycle (Deng et al. 2014).

Location of the study area, sampling sites of groundwater and sediments (star symbol), and spatial distribution of NH4–N concentrations. Vertical sampling sites of sediments along the drilling profile is marked by red rectangles
Fifty groundwater samples were collected from the resident hand tube wells within the study area. Two river water samples were included for comparative study of spectroscopy and chemistry between surface water and groundwater (Fig. 1). Groundwater was collected after a 15-min well purging. River water was filtered using a glass syringe with a 0.45-μm membrane on the site, while groundwater was filtered in an argon-gas glove box after the samples were shipped to the laboratory. Both water samples for the DOC and DOM analyses were acidized with ultra-pure HCl to pH 2 and collected in amber glass bottles to prevent sunlight. A borehole was drilled near the center of the interstream field by using PowerProbe 9500-VTR (AMS Inc., USA). Sediment samples along the profile were collected at different intervals at depths from 6 to 22.5 m belowground. Twenty-one sediment samples with seven-segement depths (three samples for each depth) (Fig. 1) were selected for subsequent DOM extraction experiment. These procedures were performed to ensure that the collected sediments demonstrated the entire properties of aquifer materials that were in contact with shallow groundwater.
Extraction of SOM
The 0.01 M CaCl2 extraction is a mild operation that is extensively performed to extract soluble organic matters in soil solutions (Toosi et al. 2012; Zsolnay 2003). We employed this technique and assumed that the extracted OM could reflect the DOM equilibrated in pore water and represent the portion that was released from the sediment into the groundwater.
The extraction procedures were conducted based on a previous study (Kalbitz et al. 2003; Toosi et al. 2012) but under an anaerobic condition. Fresh sediment samples and N2-purged 0.01 M CaCl2 solutions were mixed into centrifugation tubes at a soil–solution ratio of 1:2 (w/v) in an anaerobic glove box under purified argon flow (<0.1 ppm O2 and H2O). Sealed centrifugation tubes were then shaken for 15 min at 110 rpm under room temperature. The extracted mixtures were centrifuged at 8000×g for 30 min, transferred back into the glove box, and then filtered with 0.45-μm Teflon syringe filters. Extraction experiments were conducted in triplicate (7 × 3), and thus, 21 sediment-extracted OM (SEOM) solutions were obtained.
Spectroscopic measurements
Fluorescence excitation–emission matrices (EEM) were measured using a fluorescence spectrometer (Hitachi F-7000, Japan) for all water samples and SEOM solutions. Emission scan was conducted from a wavelength range of 300–550 nm in 2-nm increments at a stepwise increase of 4 nm with an excitation wavelength range of 250–400 nm. A 290-nm cutoff filter was used to eliminate second-order Rayleigh scattering. High absorbance samples were measured after dilutions in anaerobic chamber until the UV250 values were below 0.05 to minimize inner filter effects. All samples in a capped cuvette were measured in the previously mentioned glove box to avoid oxidation. The fluorescence signatures were blank subtracted using Milli-Q water from the original EEM. Fluorescence intensities were then normalized to the area under the water Raman peak. Instrument-specific excitation and emission corrections were applied to all spectra.
HIX was calculated following a suggested procedure (Zsolnay et al. 1999), which is based on the ratio of the emission scanning areas at a wavelength range of 300–345 to 435–480 nm with an excitation wavelength of 254 nm. High HIX values are associated with high C/H ratios, and thus have been used to indicate the humic character or autochthonous DOM component (Senesi et al. 1991; Zsolnay et al. 1999). The fluorescent BIX was determined by the ratio of the intensity at emissions of 380 and 430 nm at 310-nm excitation (Huguet et al. 2009). BIX allows an estimation of autochthonous biological activity in aquatic environments. The aromaticity indicator SUVA254 reflecting aromaticity and unsaturation of DOM (Weishaar et al. 2003; Rosario-Ortiz et al. 2007) was estimated using 254 inverse meters normalized to DOC concentration in milligrams per liter. The spectra slope (S275–295) over 275–295 nm was determined by fitting the spectral data to an exponential model (Helms et al. 2013). This index can be employed to reflect molecular weight distributions of DOM. A decrease in S275-295 suggests an increase in molecular weight (Helms et al. 2008; Wang et al. 2014). The more detailed explanations of HIX, BIX, SUVA254, and S275–295 can be referred in the cited references.
Hydrochemical analyses
Physicochemical parameters of water samples, including temperature (T), electric conductivity (EC), pH, oxidation and reduction potential (ORP), and dissolved oxygen (DO), were measured on site using a mutiparameter handheld meter (Multi 3400, Germany). NH4 + was also measured on the spot via Nessler’s reagent method with a HACH DR 2800 spectrophotometer. Major cations and anions were determined by an inductively coupled plasma atomic emission spectrometer (Thermo Electron, USA) and an ion chromatograph (Metrohm, Switzerland), respectively. Alkalinity was measured via titration. The charge balance errors of the inorganic ions were within 5 % for all samples.
The DOC and total nitrogen concentrations of water samples and SEOM solutions were determined by the high-temperature catalytic combustion method using a Shimadzu TOC-VCPH total organic carbon analyzer (Shimadzu, Co., Japan). Samples were acidified to pH <2 with HCl and were purged with CO2-free air to remove inorganic C before measurements. Each sample was measured in triplicate and reported as mean. NH4 + concentrations of SEOM solutions were determined with the same methods as those used for water samples.
Data processing and analysis
Parallel factor analysis (PARAFAC) was conducted to decompose overlapped fluorophore signatures to separate peaks. PARAFAC method was described elsewhere in detail (Bro 1997; Stedmon et al. 2003) and performed using MATLAB 8.0 (Mathworks, USA). A total of 73 EEMs, including those for 52 water samples and 21 SEOM solutions, were incorporated into the modeling process. The established model was validated by split-half and residual analyses (Stedmon et al. 2003). PARAFAC components were reported as relative concentrations represented by maximum fluorescence intensity Fmax (R.U.) or relative abundance (%). Relative abundance was calculated as the percentage of the fluorescence intensity of each component divided by the sum of the intensities of all derived components.
R-mode factor analysis (FA) was employed to identify the dominant hydrochemical variables responsible for groundwater chemistry (Güler et al. 2012; Li et al. 2014), which facilitated the understanding of the effects of NH4 + occurrence. A total of 24 variables, including groundwater physicochemical parameters, fluorescence intensity of DOM components (C1, C2, and C3), and spectroscopic parameters (SUVA254, S275–295, HIX, and BIX), were used for FA. The saturation indices (SICalcite, SIDolomite) and free-CO2 concentration (CO2f) were calculated using hydrogeochemical code PHREQC and incorporated into the dataset for FA to consider the effects of the dissolution–precipitation of carbonates. Pearson correlation of NH4 + with FOCs and the significance were calculated for both groundwater and SEOM samples. In addition to that for seven samples in the triplicate condition of the extraction experiment, the significance of correlation analysis was also given for the original 21 SEOM samples in supplementary material. FA, correlation analysis, and calculation of descriptive statistics were performed with SPSS 17.0.252.
Results
N species and groundwater properties
Physicochemical measurements for groundwater and surface water are presented in Table S1. All groundwater samples were determined with NH4–N concentrations ranging from 0.35 to 7.4 mg/L (mean 2.4 ± 1.5 mg/L), which indicates ubiquitous occurrence. About 74 % of groundwater samples (37 in 50) exhibited NH4–N concentrations below 3 mg/L. Extremely high concentration of NH4–N was not observed. This finding reveals a relatively consistent concentration distribution (Fig. 2a). Nitrate was not detectable except for only two samples with low concentrations of 0.3 and 0.82 mg/L, respectively (Table S1). The total nitrogen concentration determined was 0.41 to 8.0 mg/L (Table S1). This concentration value is close to that of NH4–N although different measurement methods were conducted. Nitrite was measured by spectrophotometry [Hatch method 10207], but it was undetectable because of its low absorbance. Groundwater DOC concentrations (mean 2.5 ± 0.6 mg/L) exhibited a more centralized distribution compared with NH4–N (Fig. 2b). A moderate correlation (r = 0.65, p < 0.01) was found between NH4–N and DOC (Fig. 2c). One surface water sample was determined with similar concentrations of NH4–N (2.75 mg/L) and TN (3.29 mg/L) to groundwater but higher DOC/NH4–N than groundwater (Fig. 2c). Both surface waters had high concentrations of nitrate over 30 mg/L (Table S1).

Concentrations of NH4–N (a) and DOC (b) in groundwater samples. Relationship between NH4–N and DOC (c) and major ion chemistry with groundwater samples displayed in black line and two surface water samples in red line (d)
HCO3 − and Ca2+ were the dominant major anion and cation, with concentrations ranging between 474 and 832 mg/L and 103 and 193 mg/L, respectively (Table S1, Fig. 2d). The concentrations of Cl−, SO4 2−, and NO3 − were consistently low and/or undetectable without exception of high concentrations (Fig. 2d). This result reveals a reducing and/or relatively closed environment of the local aquifer characterized by a lack of ion recharge. The reducing condition was also indicated by consistent negative ORP values and the significant amount of Fe2+ in the groundwater (Table S1). Two surface water samples showed evidently different ion chemistry with lower Ca2+, Mg2+, and HCO3 − and higher Cl−, SO4 2−, and NO3 − concentrations compared with the groundwater (Table S1 and Fig. 2d), which suggests anthropogenic influences.
PARAFAC components and relationships with ammonium in groundwater and SEOM solution
PARAFAC analysis showed three successfully decomposed FOCs (Fig. 3a–c). The validated split-analysis results are shown in Fig. S1. Several prior studies suggested that a three-component PARAFAC model sufficiently described all the fluorescence features of HS and/or DOM (Hur et al. 2014; Lee et al. 2015). The C1 component had a max Ex/Em of 250 (330–340)/440 nm, which was considered as fulvic-acid like component (FALC) (Fellman et al. 2010). The C2 component peaked at Ex/Em of ≤250(320)/390 nm with similar EEM peak range to humic-like materials (Coble 1996). Its fluorescent signature exhibited a slightly shorter Ex/Em, which could also be regarded as the blue shift peak position of FALC C1. The C3 component with a max Ex/Em of 275/340 nm was identified as a tryptophan-like fluorescence, suggesting amino acids or those free or bound in proteins, which indicate intact proteins or less degraded peptide material (Fellman et al. 2010).

Spectral characteristics of three PARAFAC components and the relationships of NH4–N with each component in groundwater (row 2) and SEOM solutions (row 3)
An evidently stronger correlation of NH4–N with FALC C1 (r = 0.81, p < 0.001, Fig. 3d) than that with bulk DOC (r = 0.65, p < 0.001, Fig. 2c) was observed for groundwater samples. The relationship with blue-shift FALC C2 (r = 0.79, p < 0.01, Fig. 3e) was slightly weaker but still significantly stronger than that with DOC. However, the positive relationship of NH4–N with tryptophan-like C3 decreased relative to that with DOC. The respective stronger and weaker relationships of NH4–N with FALC and tryptophan-like component C3 can be generally assumed as the two end-members of OM signature decomposed from the original moderate-correlation signature of NH4–N with DOC. By contrast, for SEOM solutions, NH4–N exhibited stronger positive correlations with FOC C1-C3 (Fig. 3g–i). The correlation trend of NH4–N with FALC C1 and that with FALC C2 in SEOM solutions was observed to be a little dissimilar to that in groundwater. Since the statistical correlations of NH4–N with FOC C1–C3 were at strong significance levels (p ≤ 0.001) both for SEOM and groundwater samples (Table S2), it allows us to make comparisons of the correlations between the different systems. Our following discussion will focus on variations of correlation levels of NH4–N with FOCs between sediments and groundwater other than the small difference among FOCs in either groundwater or SEOM solutions.
FA factors
Five varimax rotated factors were extracted according to the Kaiser criterion, accounting for 79.2 % of the total variance (Table 1). High positive loadings of factor 1 (F1) (representative parameters: C1, TN, C2, K+, NH4–N, DOC, and C3) explained the greatest amount of variance with 34.8 %, which highlights OM contribution to the groundwater chemistry (Huang et al. 2012). FOC C1–C3 incorporated into this group further demonstrated detailed indicators with specific component information for OM effects. K+ was also grouped because of the strong correlation with NH4 + in which the geochemical implication is beyond the scope of this study. Factor 2 (F2) (significant variables: EC, HCO3, Ca2+, Mg2+, and Na+), which accounted for 20.9 % of the variance, was about major ion chemistry related to water–rock interactions. Parameters of pH, SICacilate, SIDlomite, and CO2f with 10.2 % variance for factor 3 (F3) presented the sensitive factors for carbonate dissolution equilibrium. F4 (e.g., O2 and SO4 2−) and F5 (e.g., Fe2+ and ORP) clearly indicated redox effects that influenced groundwater chemistry. In general, the obtained factors identified well the important bio/hydro-geochemical effects and typical groups of physicochemical variables responsible for these effects. The results are expected to facilitate discussions of bio/hydro-geochemical impact for NH4 + occurrence in the groundwater.
Discussions
Potential NH4 + sources associated with OM
In reducing groundwater, NH4 + could come from an inorganic source, by the dissimilatory nitrate reduction to ammonium (DNRA). However, the DNRA may result in the concurrence of NO3 − and NH4 + to some extent (Hinkle and Tesoriero 2014). Thus, the ubiquitous NH4 + in the groundwater that accompanies the absence of NO3 − suggests that the DNRA is unlikely to be the main source of NH4 + (Hinkle and Tesoriero 2014) in our groundwater. In addition, the lack of intermediate nitrogen species (i.e., NO2 −) further suggests the inability of the reduction process to produce NH4 +. Therefore, significant NH4 + sources from the reduction process of NO3 − of infiltrated surface-water should be excluded. On the contrary, NH4 +-dominant nitrogen species and DOC were ubiquitously observed in the groundwater with relatively centralized concentration distribution (Fig. 2a, b) and exhibited positive relationship (r = 0.65, p < 0.01), indicating the close association of aqueous NH4 + with OM. The stronger correlation of NH4–N with the fluorescent FALC than that with DOC suggests that the occurrence of NH4 + is associated with FALC. Despite the varying correlation levels, the positive correlations of NH4 + with fluorescent humic-like and amino acid-like components observed both for groundwater and SEOM solutions indicate that NH4 + associates with these OM components (especially FALC) at varying levels in groundwater and sediments.
In sediments/soils environments, the association of SOM and ammonium-relevant species has been established. Humic and fulvic acid are complex aromatic macromolecules with amino acids, amino sugars, peptides, and aliphatic compounds that are involved in the linkages between the aromatic groups in peats and brown coals (Demirbas et al. 2006). Amino acids and proteins comprise a large fraction of organic nitrogen in lacustrine sedimentary matter (Carstens et al. 2013) and recent soils and sediments (Demirbas et al. 2006; Friedel and Scheller 2002). NH4 + are either bound to negatively charged sites of phenolic-OH and carboxylic-COOH groups of aromatic humic acid and fulivc acid or produced by mineralization processes of amino acids (e.g., urea hydrolyses) (Reeza et al. 2009). Thus, both aromatic OMs (e.g., the C1 and C2) and amino-group complexes have close affinities with NH4 +. Our geochemical analyses of sediment nitrogen showed high portions of Kjeldahl-N (93.6–99.4 %) in sediment nitrogen pool and significant amounts of NH4–N (mean 6.7 %) (unpublished data) which probably suggested the similar associations of nitrogen with OMs as established. Therefore, the correlation of NH4 + from groundwater with FOC agrees with established relationships of nitrogen–OM in sediments and thus suggests the potential of SOM as the source of NH4 + of groundwater. The declining NH4 +-OMs relationships from the stronger in sediments (Fig. 3g–i) to the relatively weaker in groundwater (Fig. 3d–f) will be discussed further later.
Hydrogeology of our study area features a seasonal water table dynamics affected by surface water and/or (possible) the presence of surface-constructed ponds. Studies in Bangladesh with the similar hydrogeology (Harvey et al. 2002; Neumann et al. 2010) showed that recharged water from ponds induced by irrigation pumping transfers degradable organic carbon to shallow aquifers. Thus, DOC in the surface-water bodies in our study area may be a possible source to N-bearing DOM of groundwater. Hydrochemical analyses showed that, in addition to SO4 2+ and NO3 −, conservative Cl− in the groundwater remained in significantly lower concentrations (mostly below 2 mg/L) compared with that in surface water (23.30 and 64.21 mg/L, Table S1). Moreover, several samples collected from the wells at the ponds (G10, G41, and G43 in Fig. 1) did not exhibit a substantially high concentration of NH4 + (or DOC) (Fig. 1 and Table S1). Apparently, the surface infiltration of N-bearing DOM (or Cl−) into the groundwater did not readily occur at the presented aquifer condition. Significant arguments (Aggarwal et al. 2003; Datta et al. 2011; van Geen et al. 2003) on the pond-derived organic carbon sources have been presented since Harvey’s proposal (Harvey et al. 2002). Reactive transport modeling (Datta et al. 2011) considering DOM adsorption/desorption indicated that the transport time of pond-derived DOM to deep aquifers exceeds typical young groundwater ages of a few decades by thousands of years. For our study, the depletion of Cl− and NH4 +-bound OM of groundwater underlying ponds could be attributed to the retardative effect of the fine texture of clayey materials of aquitards (Fig. 1) on the transport of DOM in spite of existing flow passage between surface water and groundwater.
SOM sources of NH4 + are found for typical Quaternary aquifer systems, even the sandy aquifers. Studies conducted on a sandy aquifer (Hinkle et al. 2007) in Oregon and selected principal aquifers in 48 contiguous states in the USA (Hinkle and Tesoriero 2014) suggested that NH4 + mobilization occurs from the degradation of aquifer organic matter in most sites. Our previous study (Huang et al. 2015) demonstrated the close similarity of the spectroscopic properties of DOM between groundwater and sediments. The transformation patterns of groundwater DOM (GDOM) for various samples and the analyzed hydrochemical processes integrally qualified SOM as the source of GDOM. Therefore, our data synthetically support the source of aquifer sediments for the OM-associated NH4 + of the groundwater. However, the potential contribution of leaching DOM from the bottom sediments of surface ponds to the NH4 + at sites where the top “open-doors” of aquifers exist cannot be completely ignored, which were not detected in the current study (Huang et al. 2015).
OM biodegradation effects on NH4 +–OM concurrence
One of the most important highlights by FA is the contribution of N-associated OM to groundwater chemistry suggested by F1 parameters (TN, NH4–N, DOC, C1, and C2), which could be related to the reducing characteristic of the groundwater as suggested by the parameters in F4 and F5 (i.e., depleted dissolved oxygen and SO4 2−, negative ORP and ubiquitous Fe2+). Available electron acceptors with utilizable organic carbon (electron donors) can catalyze the biodegradation of OM and promote the prevalence of reducing condition in aquifers (Stumm and Morgan 1996). Lithologic sources of DOC, instead of those surface-derived, are important sources of electron donors in shallow groundwater (Green et al. 2008; Tesoriero and Puckett 2011). Otherwise, as stated in the previous discussion, the N-associated humic materials are typically composed of soil/sediment OM components. Therefore, TN, NH4–N, and DOC grouped with FOCs as the F1 for FA (Table 1) may indicate the involvement of the N-associated SOM in bio-redox degradation processes in the aquifer.
Several data can be drawn to evaluate the biodegradation potential of SOM. The dominant relative abundance of shorter wavelength FOCs (C3 and C2, Fig. 4) of SEOM indicates a relatively simpler structure and/or lower molecular size of SOM (Lee et al. 2015; Mostofa et al. 2013) compared with that of groundwater. The simpler, smaller molecular structure and/or microbial degradation effect were commonly used to explain the blue shifts (indicating shorter wavelengths) of fluorescence peaks (Fellman et al. 2010; Helms et al. 2013). Shorter wavelength component (i.e., C3) with microbial signatures of DOM can be linked because C3 is assigned as protein/ammonia acid-like materials. Otherwise, the more liable DOM signatures of SEOM solutions were also suggested by spectroscopic indices that include higher S275–295 (smaller molecular weight), lower SUVA254 (lower aromaticity), higher BIX, and lower HIX (strong microbial sources) (Fig. S2). The information above implies that the liable small molecular fractions available from sediments (e.g., NH4-associated protein-like fraction) can be preferentially utilized by the potential biodegradation after diffusion to groundwater, whereas the relatively refractory humic-like substances (e.g., C1 and C2) were less modified and thus tend to be preserved. The varying distributions of relative abundance of FOCs among sediments and groundwater corresponding to biodegradation labilities were consistent with the maintenance of a relatively good relationship of NH4–N and FALC (C1 and C2) and the evident decline of the relationship of NH4 with protein-like OM (C3) (Fig. 3d–j and Table S2) compared with groundwater to SEOM solution.

Relative abundance of PARAFAC components in GDOM and SEOM
The significant physicochemical parameters grouped in F2 (e.g., EC, HCO3 −, Ca2+, Mg2+) suggest that abiotic hydrogeochemical process is responsible for the occurrence of HCO3 − in groundwater, which was mainly associated with mineral weathering, that increases HCO3 − with Ca2+ and Mg2+ (Gaillardet et al. 1999). However, biotic OM degradation can also contribute to HCO3 − geneses in groundwater (Stumm and Morgan 1996). To explore the influence of OM degradation on the occurrence of NH4 + and fulvic-like OMs, the normalized concentration of HCO3 − to the sum of (Ca2++Mg2+) (in mmol/L) was adopted to reflect HCO3 − trends resulted from OM biodegradation. The accompanying increase of FALC C1 and NH4–N concentration (scaled balls) with normalized HCO3 − concentration (r = 0.68, p < 0.01) (Fig. 5a) and [H+] (r = 0.64, p < 0.01) (Fig. 5b) were both observed. The results suggested that the biodegradation of complex OM produced FALC organic compositions and NH4 + from the aquifer OM precursor and increased HCO3 − and protons in groundwater. Fulvic-like OM, HCO3 −, H+, and NH4 + are common products of various OM reactions. The macromolecular humic acids/substances can be transformed into lower molecular size acidic fulvic acids and/or other forms of organic acids through anaerobic biodegradation (Lechtenfeld 2012). The mineralization of organic nitrogen can release NH4 + through the hydrolysis effect of amino acids by hydrolytic bacteria (Koho et al. 2013). Finally, the oxidative degradation of labile organic carbon (normally the smaller molecules) produces HCO3 − or CO2. The presented relationships of FALC and NH4 + with HCO3 − and H+ indicate the couple effects of these reactions that may simultaneously occur in the aquifer.

Relationship of FAL C1 concentration with normalized HCO3 − concentration (a) and [H+] (b); ball sizes were scaled to NH4–N concentrations suggesting co-increase of NH4–N with FAL C1
Conclusions
This study is the first to investigate the concurrence of NH4 + with FOCs in the groundwater of a fluvio-lacustrine aquifer at the Central Jianhan Plain by using fluorescent spectroscopic and hydrochemical methods. Results indicated the accompanying occurrence of NH4 + with FALC in the groundwater, which were mainly associated with the biodegradation of SOM.
Nitrogen contamination of water bodies is commonly attributed to anthropogenic sources, such as sewage and agricultural and industrial wastes. However, the natural occurrence of NH4 +-N in aquifers is receiving increasing attention because of its potential high loadings for nitrogen species transformations and subsequent negative effects on health and environment. Employing 3D fluorescence spectroscopy with hydrochemical analysis allows us to link NH4 + with FOCs and groundwater physicochemical parameters. The results facilitate the interpretation on the relationship of NH4 + with specific FOC types and provide new insights into NH4 + genesis in OM-rich aquifers.
References
Aggarwal PK, Basu AR, Kulkarni KM (2003) Comment on "Arsenic Mobility and Groundwater Extraction in Bangladesh" (I). Science 300:584. doi:10.1126/science.1081789
Bro R (1997) PARAFAC. Tutorial and applications. Chemometr Intell Lab Syst 38:149–171. doi:10.1016/S0169-7439(97)00032-4
Carstens D, Lehmann MF, Hofstetter TB, Schubert CJ (2013) Amino acid nitrogen isotopic composition patterns in lacustrine sedimenting matter. Geochim Cosmochim Ac 121:328–338. doi:10.1016/j.gca.2013.07.020
Coble PG (1996) Characterization of marine and terrestrial DOM in seawater using excitation emission matrix spectroscopy. Mar Chem 51:325–346
Datta S, Neal AW, Mohajerin TJ, Ocheltree T, Rosenheim BE, White CD, Johannesson KH (2011) Perennial ponds are not an important source of water or dissolved organic matter to groundwaters with high arsenic concentrations in West Bengal, India Geophys Res Lett 38:n/a-n/a doi:10.1029/2011gl049301
Demirbas A, Kar Y, Deveci H (2006) Humic Substances and Nitrogen-Containing Compounds from Low Rank Brown Coals. Energ Source Part A 28:341–351. doi:10.1080/009083190890111
Deng Y, Li H, Wang Y, Duan Y, Gan Y (2014) Temporal variability of groundwater chemistry and relationship with water-table fluctuation in the Jianghan Plain, Central China. Procedia Earth Planet Sci 10:100–103. doi:10.1016/j.proeps.2014.08.018
Duan Y, Gan Y, Wang Y, Deng Y, Guo X, Dong C (2015) Temporal variation of groundwater level and arsenic concentration at Jianghan Plain, central China. J Geochem Explor 149:106–119. doi:10.1016/j.gexplo.2014.12.001
Erskine AD (2000) Transport of ammonium in aquifers: retardation and degradation. Q J Eng Geol Hydroge 33:161–170. doi:10.1144/qjegh.33.2.161
Fellman JB, Hood E, Spencer RGM (2010) Fluorescence spectroscopy opens new windows into dissolved organic matter dynamics in freshwater ecosystems: A review. Limnol Oceanogr 55:2452–2462. doi:10.4319/lo.2010.55.6.2452
Friedel JK, Scheller E (2002) Composition of hydrolysable amino acids in soil organic matter and soil microbial biomass. Soil Bio Biochem 34:315–325. doi:10.1016/S0038-0717(01)00185-7
Gaillardet J, Dupré B, Louvat P, Allègre CJ (1999) Global silicate weathering and CO2 consumption rates deduced from the chemistry of large rivers. Chem Geol 159:3–30. doi:10.1016/S0009-2541(99)00031-5
Gleeson J, Santos IR, Maher DT, Golsby-Smith L (2013) Groundwater–surface water exchange in a mangrove tidal creek: Evidence from natural geochemical tracers and implications for nutrient budgets. Mar Chem 156:27–37. doi:10.1016/j.marchem.2013.02.001
Glessner JJG, Roy WR (2009) Paleosols in Central Illinois as potential sources of ammonium in groundwater. Ground Water Monit R 29:56–64. doi:10.1111/j.1745-6592.2009.01257.x
Green CT et al (2008) Limited occurrence of denitrification in four shallow aquifers in agricultural areas of the United States. J Environ Qual 37:994–1009. doi:10.2134/jeq2006.0419
Güler C, Kurt MA, Alpaslan M, Akbulut C (2012) Assessment of the impact of anthropogenic activities on the groundwater hydrology and chemistry in Tarsus coastal plain (Mersin, SE Turkey) using fuzzy clustering, multivariate statistics and GIS techniques. J Hydrol 414–415:435–451. doi:10.1016/j.jhydrol.2011.11.021
Harvey CF et al (2002) Arsenic mobility and groundwater extraction in Bangladesh. Science 298:1602–1606. doi:10.1126/science.1076978
Helms JR, Stubbins A, Ritchie JD, Minor EC (2008) Absorption spectral slopes and slope ratios as indicators of molecular weight, source, and photobleaching of chromophoric dissolved organic matter. Limnol Oceanogr 53(3):955–969
Helms JR, Stubbins A, Perdue EM, Green NW, Chen H, Mopper K (2013) Photochemical bleaching of oceanic dissolved organic matter and its effect on absorption spectral slope and fluorescence. Mar Chem 155:81–91. doi:10.1016/j.marchem.2013.05.015
Hinkle SR, Tesoriero AJ (2014) Nitrogen speciation and trends, and prediction of denitrification extent, in shallow US groundwater. J Hydrol 509:343–353. doi:10.1016/j.jhydrol.2013.11.048
Hinkle SR, Böhlke JK, Duff JH, Morgan DS, Weick RJ (2007) Aquifer-scale controls on the distribution of nitrate and ammonium in ground water near La Pine, Oregon, USA. J Hydrol 333:486–503. doi:10.1016/j.jhydrol.2006.09.013
Huang S, Wang Y, Cao L, Pi K, Yu M, Even E (2012) Multidimensional spectrofluorometry characterization of dissolved organic matter in arsenic-contaminated shallow groundwater. J Environ Sci Health A 47:1446–1454. doi:10.1080/10934529.2012.672390
Huang S, Liu C, Wang Y, Zhan H (2013) Multivariate analysis of the heterogeneous geochemical processes controlling arsenic enrichment in a shallow groundwater system. J Environ Sci Health A 49:478–489. doi:10.1080/10934529.2014.854689
Huang S-b, Wang Y-x, Ma T, Tong L, Wang Y-y, Liu C-r, Zhao L (2015) Linking groundwater dissolved organic matter to sedimentary organic matter from a fluvio-lacustrine aquifer at Jianghan Plain, China by EEM-PARAFAC and hydrochemical analyses. Sci Total Environ 529:131–139. doi:10.1016/j.scitotenv.2015.05.051
Huguet A, Vacher L, Relexans S, Saubusse S, Froidefond JM, Parlanti E (2009) Properties of fluorescent dissolved organic matter in the Gironde Estuary. Org Geochem 40:706–719. doi:10.1016/j.orggeochem.2009.03.002
Hur J, Kim G (2009) Comparison of the heterogeneity within bulk sediment humic substances from a stream and reservoir via selected operational descriptors. Chemosphere 75:483–490. doi:10.1016/j.chemosphere.2008.12.056
Hur J, Lee B-M, Shin K-H (2014) Spectroscopic characterization of dissolved organic matter isolates from sediments and the association with phenanthrene binding affinity. Chemosphere 111:450–457. doi:10.1016/j.chemosphere.2014.04.018
Jiao JJ, Wang Y, Cherry JA, Wang X, Zhi B, Du H, Wen D (2010) Abnormally high ammonium of natural origin in a coastal aquifer-aquitard system in the Pearl River Delta. China Environ Sci Technol 44:7470–7475. doi:10.1021/es1021697
Kalbitz K et al (2003) Changes in properties of soil-derived dissolved organic matter induced by biodegradation. Soil Bio Biochem 35:1129–1142. doi:10.1016/S0038-0717(03)00165-2
Koho KA et al (2013) Microbial bioavailability regulates organic matter preservation in marine sediments. Biogeosciences 10:1131–1141. doi:10.5194/bg-10-1131-2013
Lechtenfeld OJ (2012) Biogeochemistry of Marine Dissolved Organic Matter: Molecular Composition, Recativity and New Methods
Lee B-M, Seo Y-S, Hur J (2015) Investigation of adsorptive fractionation of humic acid on graphene oxide using fluorescence EEM-PARAFAC. Water Res 73:242–251. doi:10.1016/j.watres.2015.01.020
Li J, Wang Y, Guo W, Xie X, Zhang L, Liu Y, Kong S (2014) Iodine mobilization in groundwater system at Datong basin, China: Evidence from hydrochemistry and fluorescence characteristics. Sci Total Environ 468–469:738–745. doi:10.1016/j.scitotenv.2013.08.092
Manning DAC, Hutcheon IE (2004) Distribution and mineralogical controls on ammonium in deep groundwaters. Appl Geochem 19:1495–1503. doi:10.1016/j.apgeochem.2004.01.019
Mostofa KG, C-q L, Yoshioka T, Vione D, Zhang Y, Sakugawa H (2013) Fluorescent dissolved organic matter in natural waters. In: Mostofa KMG, Yoshioka T, Mottaleb A, Vione D (eds) Photobiogeochemistry of Organic Matter. Environmental Science and Engineering. Springer, Berlin Heidelberg, pp 429–559. doi:10.1007/978-3-642-32223-5_6
Neumann RB, Ashfaque KN, Badruzzaman ABM, Ashraf Ali M, Shoemaker JK, Harvey CF (2010) Anthropogenic influences on groundwater arsenic concentrations in Bangladesh. Nat Geosci 3:46–52, http://www.nature.com/ngeo/journal/v3/n1/suppinfo/ngeo685_S1.html
PRC Ministry of Health and Standardization Administration (2006) Standards for drinking water quality (GB 5749–2006). China Standard Press, Beijing (in Chinese)
Reeza AA, Ahmed OH, Nik Muhamad N, Jalloh MB (2009) Reducing ammonia loss from urea by mixing with humic and fulvic acids isolated from coal. Am J Environ Sci 5:420–426
Rosario-Ortiz FL, Snyder SA, Suffet IH (2007) Characterization of dissolved organic matter in drinking water sources impacted by multiple tributaries. Water Res 41:4115–4128
Santos IR, Burnett WC, Chanton J, Mwashote B, Suryaputra IG, Dittmar T (2008) Nutrient biogeochemistry in a Gulf of Mexico subterranean estuary and groundwater-derived fluxes to the coastal ocean. Limnol Oceanogr 53:705
Senesi N, Miano TM, Provenzano MR, Brunetti G (1991) Characterization, differentiation, and classification of humic substances by fluorescence spectroscopy. Soil Sci 152:259–271
Spalding RF, Exner ME (1993) Occurrence of nitrate in groundwater—a review. J Environ Qual 22:392–402. doi:10.2134/jeq1993.00472425002200030002x
Stedmon CA, Markager S, Bro R (2003) Tracing dissolved organic matter in aquatic environments using a new approach to fluorescence spectroscopy. Mar Chem 82:239–254. doi:10.1016/s0304-4203(03)00072-0
Stumm W, Morgan JJ (1996) Aquatic chemistry: Chemical equilibria and rates in natural waters, 3rd edn. Wiley, New York
Tesoriero AJ, Puckett LJ (2011) O2 reduction and denitrification rates in shallow aquifers. Water Resour Res 47:1–17. doi:10.1029/2011wr010471
Toosi ER, Clinton PW, Beare MH, Norton DA (2012) Biodegradation of soluble organic matter as affected by land-use and soil depth. Soil Sci Soc Am J 76:1667–1677. doi:10.2136/sssaj2011.0437
Umezawa Y et al (2008) Sources of nitrate and ammonium contamination in groundwater under developing Asian megacities. Sci Total Environ 404:361–376. doi:10.1016/j.scitotenv.2008.04.021
van Geen A, Zheng Y, Stute M, Ahmed KM (2003) Comment on "Arsenic Mobility and Groundwater Extraction in Bangladesh" (II). Science 300:584. doi:10.1126/science.1081057
Wang Y, Zhang D, Shen Z, Chen J, Feng C (2014) Characterization and spacial distribution variability of chromophoric dissolved organic matter (CDOM) in the Yangtze Estuary. Chemosphere 95:353–362. doi:10.1016/j.chemosphere.2013.09.044
Weishaar JL, Aiken GR, Bergamaschi BA, Fram MS, Fujii R, Mopper K (2003) Evaluation of specific ultraviolet absorbance as an indicator of the chemical composition and reactivity of dissolved organic carbon. Environ Sci Technol 37:4702–4708. doi:10.1021/es030360x
Zsolnay Á (2003) Dissolved organic matter: artefacts, definitions, and functions. Geoderma 113:187–209. doi:10.1016/S0016-7061(02)00361-0
Zsolnay A, Baigar E, Jimenez M, Steinweg B, Saccomandi F (1999) Differentiating with fluorescence spectroscopy the sources of dissolved organic matter in soils subjected to drying. Chemosphere 38:45–50. doi:10.1016/S0045-6535(98)00166-0
Acknowledgments
Dr. Shuangbing Huang was supported by the China Scholarship Council (201408130060), a grant from the National Natural Science Foundation of China (41302187), a grant from the China Post-doctoral Science Foundation (2014M552105), the Institute of Hydrogeology and Environmental Geology, Chinese Academy of Geological Sciences (SK201303, SK201412), and the Fundamental Research Funds of Chinese Academy of Geological Sciences (YYWF201503).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Responsible editor: Philippe Garrigues
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 816 kb)
Rights and permissions
About this article

Cite this article
Huang, S., Wang, Y., Ma, T. et al. Fluorescence spectroscopy reveals accompanying occurrence of ammonium with fulvic acid-like organic matter in a fluvio-lacustrine aquifer of Jianhan Plain. Environ Sci Pollut Res 23, 8508–8517 (2016). https://doi.org/10.1007/s11356-016-6092-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-016-6092-8