
Abstract
Purpose
Current synaptic vesicle 2A (SV2A) positron emission tomography (PET) imaging agents include the nanomolar affinity probes [11C]UCB-J and [18F]UCB-H derived from the anti-epileptic drug levitaracetam (Keppra®). An industry-utilized “de-risking” approach was used to carry out initial pharmacological characterization and to assess potential next-generation candidates amenable to F-18 radiolabeling for preliminary evaluation.
Procedures
Radioligand binding methods were employed in mammalian brain homogenates to determine the SV2A affinity (Kd) and maximal binding capacity (Bmax) of [3H]UCB-J. Novel leads were then screened to identify compounds minimally with comparable binding affinities with UCB-J in order to select a F-18-labeled candidate for subsequent in vivo assessment in rat. In parallel, mammalian brain tissue section autoradiography was performed to assess specific SV2A distribution.
Results
[3H]UCB-J bound with high affinity to a single population of sites in the rat brain (Kd = 2.6 ± 0.25 nM; Bmax = 810 ± 25 fmol/mg protein) and control human cortex (Kd = 2.9 ± 0.54 nM; Bmax = 10,000 ± 640 fmol/mg protein). Distribution of specific SV2A binding was shown to be homogeneous throughout the rodent brain and primarily in gray matter regions of rodent and human brain sections. Analog screening identified MNI-1038, MNI-1126/SDM-8, and SDM-2 as having comparable binding affinities with the currently available PET ligands. Subsequent [18F]MNI-1126/[18F]SDM-8 dynamic micro-PET imaging in rats revealed in vivo uptake and accumulation in the brain with favorable kinetics. Chase studies using 30 mg/kg levetiracetam confirmed that in vivo brain uptake of [18F]MNI-1126/[18F]SDM-8 was reversible.
Conclusions
Taken together, these data suggest [18F]MNI-1126/[18F]SDM-8 (since renamed as [18F]SynVesT-1) characterized via an in vitro screening cascade provided a measurable in vivo SV2A specific signal in the rodent brain. This tracer as well as the close analog [18F]SDM-2 (since renamed as [18F]SynVesT-2) is currently undergoing further evaluation in preclinical and clinical studies.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Presynaptic secretory vesicles in all mammalian brain regions contain the ubiquitously expressed transmembrane synaptic vesicle glycoprotein 2A (SV2A) [1], where it functions as a mechanism to regulate neurotransmitter release [2,3,4,5]. It is the suggested target of the anti-epileptic drugs leviteracetam and brivarectam [6,7,8], and subsequent research has garnered interest in SV2A as a potential proxy for synaptic vesicle density in a variety of neurodegenerative disorders such as Alzheimer’s disease [9, 10].
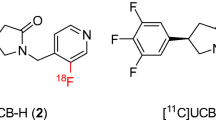
In the context of the CNS, radioligand binding studies have historically been used to quantify changes in the availability of brain proteins of interest, such as receptors, transporters, enzymes, and, more recently, misfolded proteins, in the absence and presence of novel compounds selectively targeting these proteins [11,12,13,14,15,16]. Typically, this has been achieved using standard equilibrium binding techniques [11]. Methods to rationalize and execute non-invasive in vivo assessment in a clinically translatable manner are also now fairly well understood [12]. Using positron emission tomography (PET) as the example, often the path to a clinically feasible PET radioligand requires a marriage of ligands radiolabeled with beta- and ultimately positron-emitting isotopes in order to characterize, develop and deliver a suitable agent for preclinical and clinical use [11,12,13,14,15,16]. For SV2A, initial studies led to the characterization of radiolabeled brivarecetam ([3H]UCB 34714) as a high-affinity and selective SV2A ligand in mammalian tissues [17]. More recently, efforts aimed at developing PET imaging agents for SV2A have led to the subsequent identification and development of higher affinity ligands more amenable to labeling with PET isotopes. This includes (R)-1-((3-[18F]fluoropyridin-4-yl)methyl)-4-(3,4,5-trifluorophenyl)pyrrolidin-2-one ([18F]UCB-H) and (R)-1-((3-[11C]methylpyridin-4-yl)methyl)-4-(3,4,5-trifluorophenyl)pyrrolidin-2-one ([11C]UCB-J), which have been successfully utilized for preliminary imaging studies in non-human primate and human brains [18,19,20,21]. Of these two, [11C]UCB-J is clearly superior as an imaging agent with very high levels of brain uptake and specific binding signals, while [18F]UCB-H provides significantly reduced signal to noise in comparison and will limit its utility in the clinic. This provides the need for evaluation of additional F-18-labeled analogs of UCB-J in order to facilitate widespread utility of SV2A PET imaging given the greater availability of PET centers that can produce F-18-labeled PET radioligands.
The present studies utilized an industry-adopted “de-risking” paradigm coupled to an industry/academia collaboration to investigate whether structural analogs of UCB-J had binding affinities suitable for potential development as F-18-labeled PET imaging probes for SV2A.
Materials and Methods
For details on the synthesis and characterization of the compounds as well as detailed methodologies for the studies, see the ESM.
Compounds
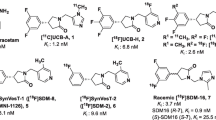
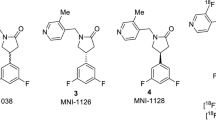
Potential SV2A ligands are shown in Fig. 1. They were synthesized based on modifications of the UCB-J structure and supplied by Invicro and the Yale PET Center.

Structures of compounds for SV2A binding assays.
Radiochemistry
Preparations of [18F]MNI-1126 for PET imaging in rodents followed the published procedures [22]. Decay-corrected radiochemical purity was > 99 %. Radiochemical yield was 8.8 % non-decay corrected and 16 % decay corrected with a specific activity of 2276 ± 96 Ci/mmol.
Homogenate Preparation
Crude brain homogenate preparations (P1) were prepared according to previously published methods [13]. The same procedure was used for human post-mortem tissue (Folio Biosciences, Miami Brain Bank, and UCSF Memory and Aging Center, USA).
In Vitro Homogenate Binding
All homogenate studies were carried out in 96-well format as previously described [14]. The IC50 values were converted to inhibition constants (Ki) to normalize for radioligand concentration in each assay using the Cheng-Prussof eqsuation [23].
Autoradiography
Autoradiography (ARG) studies were conducted as previously described [14]. In a separate series of studies, a “no-wash” assay protocol was used as described previously [15] as a gauge that a compound will potentially also develop a signal when administered in vivo [24].
In Vivo Imaging in Rat Using micro-PET
Imaging studies were conducted in Sprague-Dawley (SD) rats anesthetized with 2–3 % isoflurane (n = 4).
Results
Homogenate Binding Studies
Optimizing the Binding Protocol to Screen Novel SV2A Ligands
To provide a robust and reproducible signal, a protein concentration of 15–30 μg per well was shown to minimally provide an optimal binding window yielding a specific binding signal of 94 ± 3 % (n = 3) in rodent and human homogenates. Equilibrium was reached within 30–45 min and the percentage depletion was significantly less than 10 % (data not shown). Ligand binding to filters presoaked in 0.3 % polyetheyleneimine (PEI) was negligible (< 5 %) and not specifically displaced (data not shown).
Saturation studies revealed that [3H]UCB-J bound specifically to a single population of high-affinity sites in rat whole brain crude P1 homogenates yielding a Kd = 2.6 ± 0.25 nM and Bmax = 810 ± 25 fmol/mg protein (n = 3; Fig. 2a). The latter converts to a concentration of 41 ± 1.5 nM assuming uniform distribution and brain protein concentration of 50 mg/ml [16, 25]. Similarly in healthy control human cortex crude P1 homogenates, [3H]UCB-J bound to a single population of high-affinity sites with a Kd = 2.9 ± 0.54 nM and Bmax = 10,000 ± 640 fmol/mg protein (500 ± 32 nM, n = 7; Fig. 2b). Comparison of healthy control and AD homogenates showed an observed 57 % decrease in Bmax in the AD cortex with no significant change in binding affinity (Kd = 5.2 ± 2.1 nM and Bmax = 4300 ± 640 fmol/mg protein (215 ± 32 nM, n = 3; Fig. 2c)).

Saturation analyses of [3H]UCB-J binding to rat whole brain and human cortex crude P1 homogenates reveal binding to a single population of high-affinity sites. Data expressed as specific bound (fmol/mg protein) versus free ligand concentration (nM) for a young adult rat whole brain (n = 3), b healthy human cortex (n = 7), and c age-matched BRAAK stage IV–VI AD cortex (n = 3) homogenates. Inset shows respective Scatchard plots (bound/free versus bound (fmol/mg protein)).
Screening of Novel Potential F-18-Labeled SV2A Ligands
Assessment of compounds from Invicro and Yale was carried out in blinded fashion and included unlabeled UCB-H and UCB-J as controls. Concentration-response curves highlighting IC50’s of selected high-affinity compounds are shown in Fig. 3, comparing rat and human data with the complete datasets in Table 1. Rank ordering of the data revealed high-affinity binding (Ki ≤ 10 nM) for several of the compounds, notably MNI-1126 and MNI-1038 (Invicro) and SDM-2 and SDM-8 (Yale). MNI-1126 and SDM-8 were actually the same chemical structure (Fig. 1) and thus provided an extra internal control of the assay methodology eliciting binding affinities of < 5 nM across rat and human homogenates. All compounds had Hill slopes close to unity indicating binding to a single site. In comparison, levetiracetam (Sigma-Aldrich) generated a binding affinity of 2000 nM. The Yale compounds were provided as active and inactive enantiomers and accordingly, the latter showed lower binding affinities. Inclusion of UCB-J active enantiomer served as internal control across both studies and in which binding affinities were comparable in rat and human homogenates. Based on this information, and the availability of F-18-labeled material, MNI-1126/SDM-8 was selected for subsequent micro-PET imaging studies.

Example competition binding curves using 1 nM [3H]UCB-J in rat whole brain (a) and human cortical (b) crude P1 membranes using 10 μM UCB-H to define non-specific binding. Data is expressed as % inhibition of specific binding ± s.e.m. from 5 to 12 separate determinations carried out using 6-point concentration curves carried out in duplicate.
Autoradiography
Distribution of Specific [3H]UCB-J (1 nM) Binding in Mammalian Brain
Representative images of [3H]UCB-J (1 nM) binding showed fairly uniform and specific binding to gray matter regions throughout the rodent brain (Fig. 4; n = 3–5). Similar gray matter specific signal was observed in age-matched human healthy control and AD post-mortem cortical tissue sections (n ≥ 3). Preliminary examination and analysis suggested a decrease in signal generated in AD compared with age-matched control tissue sections (Table 2). In rTg4510 brain, a mouse model overexpressing human tau, specific [3H]UCB-J signal was also reduced compared with signal in the brains of age-matched wild-type (WT) littermates. In contrast, there was no observed change in specific signal intensity in the corresponding comparison of Tg2576 mouse brain overexpressing Aβ compared with WT littermates.

Example autoradiograms in mammalian brain sections showing distribution of [3H]UCB-J (1 nM) binding under equilibrium conditions in mammalian brain regions representing normal and diseased states. WT are littermates for each of the transgenic animal models, rTg4510 tau–overexpressing mice, or Tg2576 amyloid–overexpressing mice.
Comparison of Healthy Control and AD Sections Using a Wash and No-Wash Characterization
The observed [3H]UCB-J signal increased as expected switching from low (1 nM) to saturating (20 nM) radioligand concentration (Fig. 5) under conditions where the tissue sections were washed in buffer and dried prior to exposure.

Example autoradiograms in human brain sections showing distribution signal of [3H]UCB-J (1 and 20 nM) under equilibrium conditions in comparison with normal rat brain tissue under normal washing conditions. This is compared with autoradiograms in human brain sections showing distribution signal of [3H]UCB-J (1 and 20 nM) under “no-wash” conditions.
Using the “no-wash” protocol, [3H]UCB-J (1 nM) specific signals developed in normal healthy control cortex (n = 3–5) and hippocampal sections (n = 3; Fig. 5) of 96 ± 1.2 % and 97 ± 0.4 % respectively. Similar specific signals developed at 20 min in WT mouse and naïve rat whole brain sections, respectively (n = 3–5; not shown). These were derived from radioactivity counted on GF/B filter discs using liquid scintillation counting and in which average counts were in the range of 11,000 dpm (total) and 700 dpm (Ns). As expected, the data from the washed sections was reproduced if sections were exposed to phosphorimaging screens. Table 3, in the ESM, shows the results from the quantification of the data for all groups. These values are comparable with the tritiated version of the gold standard clinical GABA-A G protein–coupled receptor PET ligand [11C]flumazenil [24] and suggestive of a reasonable probability of success for providing a measurable in vivo signal with the caveat of sufficient delivery across the blood-brain barrier and subsequent retention at the target.
Dynamic Micro-PET Imaging in Rat Brain Using [18F]MNI-1126/[18F]SDM-8
Systemic administration of [18F]MNI1126/[18F]SDM-8 (23 ± 1 MBq, n = 4) into the tail vein of male SD rats followed by dynamic acquisition over 90 min revealed a time-activity curve profile consistent with rapid “wash-in,” and fast “washout” kinetics typical of successful in vivo CNS imaging agents (Fig. 6). Baseline scans were performed in 2 animals. To demonstrate the reversibility of [18F]MNI-1126/[18F]SDM-8 binding, a dynamic PET scan acquired over the same timecourse was employed followed by a chase. Since [18F]MNI-1126/[18F]SDM-8 reaches a maximum in vivo accumulation in the brain between 15 and 30 min (n = 4), at 30 min and while still acquiring the dynamic data, 30 mg/kg levetiracetam was administered intravenously. Here, the initial 15–30-min rapid peak to steady state was followed by a relatively faster washout of tracer compared with that in the absence of levetiracetam indicating reversibility of the in vivo binding of [18F]MNI-1126/[18F]SDM-8 in the rat brain (Fig. 6).

Micro-PET-derived time-activity curves of [18F]MNI-1126/[18F]SDM-8 in rat brain based upon 90-min dynamic acquisition protocol. Arrow indicates 30 mg/kg i.v. levetiracetam administration into the tail vein catheter during the scan (n = 3–4). Panel a shows a single animal with a preblock scan and a post blocking chase scan on a separate day. Insets show the summed images at the times indicated for the animal under both conditions. Panel B shows the mean ± SE of baseline scans (n = 2) compared with blocking scans in 4 animals.
Discussion
Changes in synaptic physiology over the course of a lifetime are thought to play an important role in the physiologic and pathologic processes regulating synaptic density [1,2,3,4]. Longitudinal quantification of synaptic densities in the living human brain is essential to the understanding, diagnosis, and treatment of brain disorders associated with synaptic pathology. Currently, such non-invasive and longitudinal in vivo clinical quantification of SV2A is achievable using [18F]UCB-H and [11C]UCB-J [19,20,21]. Development of next-generation F-18-labeled SV2A PET radioligands is warranted to improve on metrics such as in vivo signal to noise, in part to minimize inter-subject quantification error as well as leveraging the longer radioactive half-life of fluorine-18 and the wider availability of centers that can competently work with this isotope compared with the more specialist use of carbon-11. Additionally, optimizing the ease and reproducibility of radiosynthetic methods is of particular importance for delivering reliable quantities of clinical grade material required for human studies. If novel tracer candidates are available for assessment, the question remains as to which candidate(s) to select for ultimate development. As such, a data-driven approach to de-risk which candidate to select provides an efficient, cost-effective method to make go/no-go decisions in this respect.
The use of radioligand binding approaches to characterize and profile CNS protein targets and potential probes has been well characterized [11]. Likewise, the optimal physicochemical properties for successful CNS imaging radiotracers are well understood and have been documented using a de-risking approach that has been adopted and utilized by pharma to develop and deliver clinical CNS PET imaging probes [11,12,13,14,15, 26]. An initial step in this process is to establish that the candidate has high affinity for the target of interest; generally < 5 nM is a good starting point as most currently used clinical PET radiotracers fall into this range. These factors allow prediction of the likelihood of obtaining a measureable in vivo specific signal based in part on the relationship of the ratio of target density to ligand affinity (Bmax/Kd) originally proposed by Eckelmann and Gibson [26].
Using tritium-labeled material initially provides a relatively affordable means of early characterization of the candidate radioligand and/or a tool compound in a higher throughput, faster turnaround manner over a timeframe not limited by radioactive decay half-life. In these types of studies, specific binding can be determined in three potential ways: (a) by co-incubation of radioligand with tissue sample and an excess of some competing ligand. Such self (homologous) competition will provide a specific signal but runs the inherent risk of missing any potential off-target activity as radioligand and competing compound will bind to all the same known and unknown sites. Alternatively, (b) a structural analog differing in some way to the radioligand can be used to mitigate the caveat highlighted in (a). Better still, if available, is the use of (c) a structurally dissimilar compound with known selectivity for the target of interest. In the present studies, unlabeled UCB-H was utilized to assess non-specific binding satisfying approach (b) listed above. In this case, optimizing the [3H]UCB-J binding assay in mammalian brain preparations served to provide a template describing the stepwise approach that can be generally applied to any novel radioligand radiolabeled with most isotopes, and with the ultimate goal of providing the information necessary to rank-order the test compounds for affinity. In addition, knowing the Bmax of the target allows estimation of the Bmax/Kd ratio for the ultimate PET ligand candidate that provides a decision-making opportunity on whether to trigger and/or continue development on satisfying the Eckelmann/Gibson index of ≥ 10 [26]. As an example, the Bmax/Kd ratio for [3H]UCB-J in rodent and human brains assessed in these studies is 16 and 170, respectively, and supports the published in vivo data in humans and non-human primates to date [18,19,20,21]. Interestingly, saturation analysis in crude P1 homogenates prepared from the post-mortem AD cortex in these studies revealed a 57 % reduction in Bmax that correlated with the reduction observed in the tissue ARG studies and consistent with the magnitude of reduction (41 % decrease in nondisplaceable binding potential, BPND, in the hippocampus of patients with Alzheimer’s disease) measured in vivo with [11C]UCB-J PET imaging [27].
Parallel ARG studies executed in rodent sagittal brain sections confirmed a fairly uniform distribution across mammalian brain consistent with published in vivo data [18, 19], with higher levels of specific binding observed in gray matter of the mammalian cortex and hippocampus. Post-mortem human tissue sections revealed a similar propensity for the specific signal occurring in gray, compared with white matter regions of the cortex and hippocampus sections assessed. In the absence of any existing data, typically this approach would use antibody immunostaining (IHC) protocols on adjacent sections to confirm that the distribution of specific radiochemical signal matched that provided by the IHC methods. When stock radioligand and/or tissue sample availability is limited, another option is to run the ARG method using a saturating concentration of radioligand. This can be achieved using a radioligand concentration approximately tenfold higher than the Kd of the radioligand for its target. The information provided in this way can then be used to estimate changes in target density that may occur between healthy and diseased tissue. Accordingly, at a sub-saturating (1 nM) concentration of [3H]UCB-J, a reduction in the specific binding signal was observed in the tau-overexpressing rTg4510 mouse brain and human AD post-mortem cortex and hippocampus. In contrast, no change in SV2A signal was observed in the Aβ-overexpressing Tg2576 mouse brain. This may be linked to the somewhat equivocal data citing the extent of neuronal loss at the age of the mice used in this study, and indicative of a lack of synaptic loss in this particular cohort of animals [28, 29]. A similar observation was noted by running the studies at the saturating (20 nM) radioligand concentration and further supported in the cortical homogenate studies where detailed saturation curves were generated. Of course context is key since an artificial increase in signal to noise established by the wash paradigm in the in vitro methods is absent in in vivo PET studies.
We have previously demonstrated that by using a “no-wash protocol,” the tritiated version of the “gold standard” clinical CNS PET radioligand [11C]flumazenil provided an approximate 95 % specific binding signal under the conditions employed [14]. In contrast, the tritiated version of the dopamine D2 receptor radioligand [11C]raclopride provided an approximate 30 % specific binding signal. Using these as higher and lower cutoff points respectively, it was shown that candidates tested that fall between these two values subsequently provided measurable in vivo signals when administered as PET-labeled versions in rodents and humans [14, 25]. In the present studies [3H]UCB-J elicited a signal close to that of radiolabeled flumazenil in both human and rodent tissues thus suggesting that radiolabeled UCB-J should provide a measurable in vivo signal as has indeed been reported [19, 20].
Armed with this in vitro data package, preliminary in vivo assessment of [18F]MNI-1126/[18F]SDM-8 in rat using micro-PET imaging could be triggered with confidence. For a viable PET tracer, following the uptake phase, there should be a distribution pattern that reflects the binding site density. Other notable characteristics include reversibility of the tracer by displacement with a structurally distinct compound. For [18F]MNI-1126/SDM-8, the tracer was shown to rapidly enter the brain and establish a plateau level from 15 to 30 min followed by washout phase at a slower rate than uptake. A bolus i.v. injection of the SV2A ligand levetiracetam (30 mg/kg) at 30 min resulted in a faster reduction in signal indicative of levetiracetam competing for binding and thus showing reversibility of the binding interaction in vivo [25]. Furthermore, the difference between the normal washout curve in the presence and absence of competing ligand can be used to estimate the size of the signal expected in future in vivo target occupancy studies. Given that [18F]MNI-1126/[18F]SDM-8 displayed relatively slow kinetics in NHPs [22], it is likely to observe even slower kinetics in humans though this remains to be seen. This is obviously another factor to consider when de-risking potential clinical PET ligand probes. These in vivo data thus suggest that [18F]MNI-1126/[18F]SDM-8 is a viable tracer for monitoring SV2A in rodent models. Results from studies in non-human primates have since been published and confirmed the suitability of [18F]MNI-1126/[18F]SDM-8 to image SV2A and provide high levels of specific binding signals [22, 30].
Conclusions
In summary, in vitro radioligand binding and in vivo micro-PET imaging methods have been utilized to characterize several SV2A-targeting small molecules in order to provide data-driven decisions on which analog(s) to pursue as potential F-18-labeled clinical PET ligands for SV2A. Here, [18F]MNI-1126/[18F]SDM-8, recently renamed as [18F]SynVesT-1, has emerged as a promising candidate. Assessment for this tracer as well as for the equally promising candidate [18F]SDM-2 (renamed [18F]SynVesT-2) in non-human primates and humans is the subject of other studies.
References
Bajjalieh SM, Frantz GD, Weimann JM, McConnell S, Scheller RH (1994) Differential expression of synaptic vesicle protein 2 (SV2) isoforms. J Neurosci 14:5223–5235
Crowder KM, Gunther JM, Jones TA, Hale BD, Zhang HZ, Peterson MR, Scheller RH, Chavkin C, Bajjalieh SM (1999) Abnormal neurotransmission in mice lacking synaptic vesicle protein 2A (SV2A). Proc Natl Acad Sci 96:15268–15273
Jahn R, Fasshauer D (2012) Molecular machines governing exocytosis of synaptic vesicles. Nature 490:201–207
Mendoza-Torreblanca JG, Vanoye-Carlo A, Phillips-Farfán BV et al (2013) Synaptic vesicle protein 2A: basic facts and role in synaptic function. Eur J Neurosci 38:3529–3539
Vogl C, Tanifuji S, Danis B, Daniels V, Foerch P, Wolff C, Whalley BJ, Mochida S, Stephens GJ (2015) Synaptic vesicle glycoprotein 2A modulates vesicular release and calcium channel function at peripheral sympathetic synapses. Eur J Neurosci 41:398–409
Lynch BA, Lambeng N, Nocka K, Kensel-Hammes P, Bajjalieh SM, Matagne A, Fuks B (2004) The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc Natl Acad Sci U S A 101:9861–9866
Kenda BM, Matagne AC, Talaga PE, Pasau PM, Differding E, Lallemand BI, Frycia AM, Moureau FG, Klitgaard HV, Gillard MR, Fuks B, Michel P (2004) Discovery of 4-substituted pyrrolidone butanamides as new agents with significant antiepileptic activity. J Med Chem 47:530–549
von Rosenstiel P (2007) Brivaracetam (UCB 34714). Neurotherapeutics 4(1):84–87
Ohno Y, Tokudome K (2017) Therapeutic role of synaptic vesicle glycoprotein 2A (SV2A) in modulating epileptogenisis. CNS Neurol Disord Drug Targets 16:463–471
Löscher W, Gillard M, Sands ZA, Kaminski RM, Klitgaard H (2016) Synaptic vesicle glycoprotein 2A ligands in the treatment of epilepsy and beyond. CNS Drugs 30:1055–1077
Lever SZ, Fan KH, Lever JR (2017) Tactics for preclinical validation of receptor-binding radiotracers. Nucl Med Biol 44:4–30
Hargreaves RJ, Hoppin J, Sevigny J, Patel S, Chiao P, Klimas M, Verma A (2015) Optimizing central nervous system drug development using molecular imaging. Clin Pharmacol Ther 98:47–60
Patel S, Hamill TG, Connolly B, Jagoda E, Li W, Gibson RE (2007) Species differences in mGluR5 binding sites in mammalian central nervous system determined using in vitro binding with [18F]F-PEB. Nucl Med Biol 34:1009–1017
Patel S, Ndubizu O, Hamill T, Chaudhary A, Burns HD, Hargreaves R, Gibson RE (2005) Screening cascade and development of potential positron emission tomography radiotracers for mGluR5: in vitro and in vivo characterization. Mol Imaging Biol 7:314–323
Patel S, Hammil T, Hostetler E et al (2003) An invitro assay for predicting successful imaging radiotracers. Mol Imaging Biol 5:65–71
Patel S, Gibson R (2008) In vivo site-directed radiotracers: a mini-review. Nucl Med Biol 35:805–815
Gillard M, Fuks B, Leclercq K, Matagne A (2011) Binding characteristics of brivaracetam, a selective, high affinity SV2A ligand in rat, mouse and human brain: relationship to anti-convulsant properties. Eur J Pharmacol 664:36–44
Warnier C, Lemaire C, Becker G, Zaragoza G, Giacomelli F, Aerts J, Otabashi M, Bahri MA, Mercier J, Plenevaux A, Luxen A (2016) Enabling efficient positron emission tomography (PET) imaging of synaptic vesicle glycoprotein 2A (SV2A) with a robust and one-step adiosynthesis of a highly potent 18F-labeled ligand ([18F]UCB-H). J Med Chem 59:8955–8966
Nabulsi NB, Mercier J, Holden D, Carre S, Najafzadeh S, Vandergeten MC, Lin SF, Deo A, Price N, Wood M, Lara-Jaime T, Montel F, Laruelle M, Carson RE, Hannestad J, Huang Y (2016) Synthesis and preclinical evaluation of 11C-UCB-J as a PET tracer for imaging the synaptic vesicle glycoprotein 2A in the brain. J Nucl Med 57:777–784
Finnema SJ, Nabulsi NB, Eid T, Detyniecki K, Lin SF, Chen MK, Dhaher R, Matuskey D, Baum E, Holden D, Spencer DD, Mercier J, Hannestad J, Huang Y, Carson RE (2016) Imaging synaptic density in the living human brain. Sci Transl Med 8:348ra96
Finnema SJ, Nabulsi NB, Mercier J, Lin SF, Chen MK, Matuskey D, Gallezot JD, Henry S, Hannestad J, Huang Y, Carson RE (2018) Kinetic evaluation and test-retest reproducibility of [11C]UCB-J, a novel radioligand for positron emission tomography imaging of synaptic vesicle glycoprotein 2A in humans. J Cereb Blood Flow Metab 38:2041–2052
Li S, Cai Z, Wu X, Holden D, Pracitto R, Kapinos M, Gao H, Labaree D, Nabulsi N, Carson RE, Huang Y (2019) Synthesis and in vivo evaluation of a novel PET radiotracer for imaging of synaptic vesicle glycoprotein 2A (SV2A) in nonhuman primates. ACS Chem Neurosci 10:1544–1554
Cheng Y, Prusoff WH (1973) Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol 22:3099–3108
Patel S, Hamill T, Hostetler E et al (2003) An in-vitro assay for predicting successful imaging radiotracers. Mol Imaging Biol 5:65–71
Nicolas JM, Hannestad J, Holden D, Kervyn S, Nabulsi N, Tytgat D, Huang Y, Chanteux H, Staelens L, Matagne A, Mathy FX, Mercier J, Stockis A, Carson RE, Klitgaard H (2016) Brivaracetam, a selective high-affinity synaptic vesicle protein 2A (SV2A) ligand with preclinical evidence of high brain permeability and fast onset of action. Epilepsia 57:201–209
Eckelman WC, Gibson RE (1993) The design of site-directed radiopharmaceuticals for use in drug discovery. In: Burns HD, Gibson RE, Dannals R, Siegl P (eds) Nuclear imaging and drug discovery, development and approval. Birkhauser, Boston, pp 114–134
Chen MK, Mecca AP, Naganawa M, Finnema SJ, Toyonaga T, Lin SF, Najafzadeh S, Ropchan J, Lu Y, McDonald JW, Michalak HR, Nabulsi NB, Arnsten AFT, Huang Y, Carson RE, van Dyck CH (2018) Assessing synaptic density in Alzheimer disease with synaptic vesicle glycoprotein 2A positron emission tomographic imaging. JAMA Neurol 75:1215–1224
Dawson TM, Golde TE, Lagier-Tourenne C (2018) Animal models of neurodegenerative diseases. Nat Neurosci 21:1370–1379
Sasaguri H, Nilsson P, Hashimoto S, Nagata K, Saito T, de Strooper B, Hardy J, Vassar R, Winblad B, Saido TC (2017) APP mouse models for Alzheimer’s disease preclinical studies. EMBO J 36:2473–2487
Constantinescu CC, Tresse C, Zheng M, Gouasmat A, Carroll VM, Mistico L, Alagille D, Sandiego CM, Papin C, Marek K, Seibyl JP, Tamagnan GD, Barret O (2019) Development and in vivo preclinical imaging of fluorine-18-labeled synaptic vesicle protein 2A (SV2A) PET tracers. Mol Imaging Biol 21:509–518
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
(PDF 341 kb)
Rights and permissions
About this article

Cite this article
Patel, S., Knight, A., Krause, S. et al. Preclinical In Vitro and In Vivo Characterization of Synaptic Vesicle 2A–Targeting Compounds Amenable to F-18 Labeling as Potential PET Radioligands for Imaging of Synapse Integrity. Mol Imaging Biol 22, 832–841 (2020). https://doi.org/10.1007/s11307-019-01428-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11307-019-01428-0