
Abstract
Ectonucleotidase inhibitors are a family of pharmacological drugs that, by selectively targeting ectonucleotidases, are essential in altering purinergic signaling pathways. The hydrolysis of extracellular nucleotides and nucleosides is carried out by these enzymes, which include ectonucleoside triphosphate diphosphohydrolases (NTPDases) and ecto-5′-nucleotidase (CD73). Ectonucleotidase inhibitors can prevent the conversion of ATP and ADP into adenosine by blocking these enzymes and reduce extracellular adenosine. These molecules are essential for purinergic signaling, which is associated with a variability of physiological and pathological processes. By modifying extracellular nucleotide metabolism and improving purinergic signaling regulation, ectonucleotide pyrophosphatase/phosphodiesterase (ENPP) inhibitors have the potential to improve cancer treatment, inflammatory management, and immune response modulation. Purinergic signaling is affected by CD73 inhibitors because they prevent AMP from being converted to adenosine. These inhibitors are useful in cancer therapy and immunotherapy because they may improve chemotherapy effectiveness and alter immune responses. Purinergic signaling is controlled by NTPDase inhibitors, which specifically target enzymes involved in extracellular nucleotide breakdown. These inhibitors show promise in reducing immunological responses, thrombosis, and inflammation, perhaps assisting in the treatment of cardiovascular and autoimmune illnesses. Alkaline phosphatase (ALP) inhibitors alter the function of enzymes involved in dephosphorylation reactions, which has an impact on a variety of biological processes. By altering the body’s phosphate levels, these inhibitors may be used to treat diseases including hyperphosphatemia and certain bone problems. This article provides a guide for researchers and clinicians looking to leverage the remedial capability of ectonucleotidase inhibitors in a variety of illness scenarios by illuminating their processes, advantages, and difficulties.
Graphical Abstract
Ectonucleotidases provide a visually appealing way to explore the complex world of extracellular nucleotide metabolism. The abstract’s central visual element is a stylized depiction of the cell membrane, emphasizing the surface-bound ectonucleotidases that are essential for controlling the amounts of extracellular nucleotides. The graphical section goes on to illustrate the wider consequences of ectonucleotidase activity, addressing a number of biological mechanisms, including the regulation of immune response and neurotransmission.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
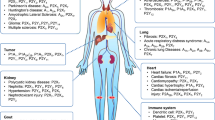
The breakdown of extracellular nucleotides (like ATP and ADP) into nucleosides (like adenosine and inorganic phosphates) is carried out by membrane-bound enzymes called ectonucleotidase enzymes. These enzymes are articulated on the cell exterior of various cell types, including the immune system, endothelial cells, and neurons. Extracellular nucleotide levels need to be regulated because, depending on the nucleotide and cell type involved, they can have both pro- and anti-inflammatory effects. For instance, pro-inflammatory cytokines can be generated when injured or stressed cells release ATP, which can then trigger inflammation. Adenosine, on the other hand, possesses immunosuppressive and anti-inflammatory properties. Dysregulation of exonucleases is correlated with various diseases such as cancer, inflammation, autoimmune diseases, and neurological diseases [1, 2]. Ectonucleotidases act a significant role in the dephosphorylation of various nucleotides and nucleosides involved in the activation of purinergic receptors (P1 and P2) (Fig. 1) [3,4,5]. P1 receptor, also known as adenosine receptor, is activated by adenosine and is a G-coupled protein receptor. Caffeine, theophylline, and other methylxanthines frequently target these receptors. Adenosine receptors are multipotent and have four subtypes: A1, A2A, A2B, and A3 [6,7,8,9]. While A1 receptors are mainly involved in the regulation of the heart and blood pressure, A2A receptors are expressed in endothelial cells, heart, fibroblasts, infiltrated hematopoietic cells and myocardial fibrils. Additionally, A2A and A2B receptors exhibit anti-inflammatory activities in organs such as the lungs, intestines, liver, and kidneys. A3 receptors are G protein–coupled receptors widely distributed in the testes, spleen, liver, and various organs in humans and other organs. They play a role in functions such as inflammation and immunity [10,11,12]. Nucleotide-activated P2 receptors are expressed on immune and non-inflammatory cells throughout the body. Based on their characteristics, P2 receptors may be classified into two groups: metabotropic P2Y receptors and ionotropic P2X receptors. An ionotropic receptor that acts as a membrane ion channel permeable to calcium, potassium, and sodium is the trimeric ATP-activated P2X receptor. P2X receptors are in seven varieties, from P2X1 to P2X7 [13, 14]. The human body has many of these receptors, particularly in the neurological and cardiovascular systems. They perform a significant function in many physiological processes such as the sense of taste, smooth muscle contraction, cough, vision loss, and neurodegenerative diseases [15, 16].

Purinergic receptor (P1 and P2) agonists
The metabotropic purinergic receptor family includes P2Y receptors, which are activated by extracellular nucleotides, specifically ATP and ADP. The eight subtypes of P2Y receptors that are known to exist are P2Y1, P2Y2, P2Y4, P2Y6, and P2Y11-P2Y14. These subtypes are found throughout the body and serve different purposes. P2Y1 receptors are mostly located in the cardiovascular system, where they are involved in the formation of thrombi and platelet aggregation [17]. P2Y2 receptors are expressed in several tissues, such as the respiratory, urinary, and gastrointestinal tracts, where they contribute to fluid secretion and inflammation [18]. P2Y4 receptors are primarily expressed in the digestive and respiratory systems, where they are involved in mucin secretion and inflammation [19]. P2Y6 receptors are associated with immunity and inflammation, particularly the activation of microglia and stellate cells in the CNS [20]. P2Y11 receptors are mainly found in the gastrointestinal tract and contribute to muscle contraction and the bladder [21]. The P2Y12 receptor is expressed only on platelets, participates in thrombosis, and is a target of anti-platelet drugs such as clopidogrel [22]. The P2Y13 receptor is also present on platelets and plays a role in platelet activation and aggregation [23]. The P2Y14 receptor is expressed in several tissues, including the immune system, and performs a function in inflammation and cell migration. The most common agonists of P2Y receptors are ATP, ADP, UTP, and UDP (Fig. 2) [24, 25].

Classification of purinergic receptors
Ectonucleotidases, types, functions, and importance of inhibition
Ectonucleotide pyrophosphatase/phosphodiesterases (ENPPs), ectonucleoside triphosphate diphosphohydrolases (NTPDases), alkaline phosphatases (ALP), and ecto-5′-nucleotidase (CD73) are the four different forms of ectonucleotidases.
Ectonucleotide pyrophosphatase/phosphodiesterases (ENPPs)
The ectonucleotide pyrophosphatase/phosphodiesterase (ENPP) family consists of seven enzymes that are important in regulating extracellular nucleotide and nucleoside concentrations in a kind of physiological processes, such as bone mineralization, inflammation, and cardiac function. Among these enzymes, ENPP1 and ENPP2 are the most studied. Transmembrane glycoprotein ENPP1 is implicated in the pathophysiology of ectopic calcification, type 2 diabetes, and insulin resistance by interfering with insulin control [26, 27]. Often referred to as autocrine motility factor, ENPP2 is a secreted enzyme that converts LPC, a lipid mediator involved in cellular activities such cell migration, proliferation, and survival, into LPA. Moreover, inflammation, cancer, and heart disease are associated with ENPP2. ENPP3, also known as CD203c, is a transmembrane glycoprotein widely expressed in the immune system. Finally, ENPP6 is a transmembrane glycoprotein expressed in tissues such as bone, cartilage, and fat, regulating bone mineralization, insulin signaling, and inflammation (Fig. 3) [28,29,30].

Function of ENPPs and its inhibitions
Ecto-5′-nucleotidase (CD73)
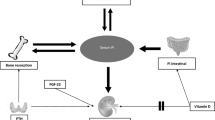
The glycoprotein known as ecto-5′-nucleotidase, or CD73, is found on the cell membrane. It is present in several organs, including the immune system, cell endothelial inflammation, and cancer cells. Its activity and function are controlled by a variety of variables, including hypoxia, growth factors, and cytokines [29]. By blocking adenosine signaling, CD73 regulates immunological response, blood flow, and tissue healing. Furthermore, a variety of illnesses, such as cancer, inflammatory disorders, and cardiovascular diseases, have been linked to dysregulation of CD73 expression and activity. Therefore, CD73 is a promising therapeutic target, and CD73 inhibitors have been shown in previous studies to be anti-tumor, anti-cancer, and anti-inflammatory. It has been shown that adenosine signaling agonists can treat heart disease (Fig. 4) [31,32,33,34,35,36,37].

Ecto-5ʹ-nucleotidase function in catalysis of natural substrate AMP
Ectonucleoside triphosphate diphosphohydrolases (NTPDases)
A family of enzymes known as nucleoside triphosphate diphosphate hydrolase (NTPDase) hydrolyzes the strength of extracellular nucleotides, including ATP and ADP, to regulate them. The eight members of the NTPDase family have a role in physiological processes like immunological response, nervous system function, and platelet aggregation [1]. They are expressed in an array of organs and cell classes. Diseases including cancer, inflammatory illnesses, and cardiovascular disease have all been linked to dysregulation of NTPDase expression and activity [31]. NTPDase1, also known as CD39, is the most studied member of this family and performs a significant function in regulating the immune system and platelet function. Other members, such as NTPDase3 and NTPDase5, have been shown to perform a function in cancer progression and may serve as therapeutic targets. NTPDase inhibitors, including CD39, show promise for the therapy of diseases such as cancer and inflammatory diseases (Fig. 5) [38,39,40,41,42].

Function of NTPDases
Alkaline phosphatases (APs)
Alkaline phosphatase (ALP) is an enzyme that works by hydrolyzing phosphate esters under physiological pH. They are expressed in many tissues and cell types, including bone, liver, and intestine, and are encoded by four human-known genes, ALPL, ALPP, ALPI, and ALPG. ALP has many physiological effects, such as bone mineralization, liver function, and intestinal function [43, 44]. They play a significant function in the synthesis and formation of bone matrix in bones, and in the liver, ALP is exploited as a indicator of liver disorder and its activity increases in cholestasis. They participate in the dephosphorylation of dietary phosphates in the intestine and are important for their absorption [45]. Changes in ALP activity are correlated with many disorders such as osteoporosis, liver disease, and cancer. Changes in the ALPL gene, responsible for encrypting tissue-non-specific ALP (TNAP), result in hypophosphatasia, an uncommon condition characterized by bone loss [46, 47]. High ALP activity in liver disease is diagnostic of cholestasis, while decreased ALP activity is associated with liver fibrosis. Increased ALP activity is associated with poor diagnosis in many types of cancer. ALP inhibitors have been studied for the treatment of many diseases. For example, ALP inhibitors are being explored for use in the remedy of osteoporosis and cancer bone metastases. ALP inhibitors have also been proposed for the treatment of liver fibrosis (Fig. 6) [48,49,50].

Structure of phosphorylated prodrug and ALP
Inhibitor of ectonucleotide pyrophosphatase/phosphodiesterases (ENPPs)
Nucleotide-based inhibitors
Eliahu et al. reported diadenosine polyphosphonate derivatives 1 evaluated as ENPP inhibitors. All analogs inhibited the catabolism of pnp-TMP (Ki and IC50 were determined to be between 10 and 60 μM), Ap5A, and ATP by ENPP1 and prevented over 80% of the ENPP2-dependent hydrolysis of pnp-TMP, a particular ENPP substrate. The novel analogs suppressed ENPP3 activity to a lesser amount; compounds 1a and 1d were the most effective in this regard. These analogs reduce pnp-TMP hydrolysis levels in bone and colon cancer cells. Significantly, derivatives 1a–1d exhibited reduced activity at human P2Y1,11 receptors (excluding derivative 1a) but no action at human P2Y2 receptors. These findings offer compelling proof that analog 1b is the first unique ENPP inhibitor discovered [51]. ENPP1 inhibitors based on uridine dithiophosphate derivatives 2–5 were described by Zelikman et al. Entirely, these derivatives can inhibit h-ENPP1 at 100 μM (80–100% inhibition), while ENPP3 and other ectonucleotidases (NTPDase1, 2, 3, 8) have no or very little inhibition. These compounds relate to selective ENPP1 inhibitors because of their moderate effect at the uracil nucleotide–sensitive P2Y2,4,6-receptor. With a Ki value of 27 nM, diuridine 5 are the most effective inhibitors. Derivatives 2–5 have been shown to be stable in acidic or alkaline pH as well as resistant to atmospheric oxidation. MDS have shown that the improved ENPP1 repressing action and selectivity of derivative 5 can be recognized to material simultaneously occupying two regions of ENPP1 (AMP region and other regions) (Fig. 7) [52]. Nadel and associates synthesized six different forms of ATP-α-SH-β, including γ-methylene (6a), ATP-α-SH-β, γ-dichloromethyl (6b), ATP-α-methylene-γ-SH (6c), and eight-thio-ATP (6d), and have shown how they conflicted with hydration by ENPP1,3 and NTPDase1, 2, 3, 8 (< 5% hydrolysis). The hydrolysis of thymidine analogs by ENPP1 and ENPP3 was inhibited by derivatives 6a–6c by > 90% and 23–43%, respectively, at 100 μM, whereas NTPDase1, 2, 3, 8 hydrolysis was only marginally impacted (0–40%). With Ki = 20 nM and IC50 = 0.39 μM, derivative 6c is the strongest ENPP1 inhibitor discovered thus far. With a Ki of 685 nM, derivative 6b is a selective inhibitor of ENPP1 showing an IC50 of 0.57 μM. It has been demonstrated that derivatives 6a–6c are specifically non-agonists of P2Y1/P2Y2/P2Y11 receptors. MDS of 6a–6c derivatives into the ENPP1 model showed that the activity is related to the binding site and the number of hydrogen bonds to residues. Briefly, analogs 6b and 6c are excellent inhibitors of ENPP1 [53]. Lecka et al. narrated the creation of 13 non-hydrolyzable ATP derivatives 7 and evaluated as selective h-ENPP1 inhibitors. The hydrolysis of pnp-TMP by recombinant ENPP1 and cell surface ENPP1 action in bone tumor cells (HTB-85) was reduced (66–99%) by all derivatives at 100 μM. The activity of ENPP3 and NTPDase, the other ectonucleotidases, is only marginally affected by these derivatives. With Ki,app values extending from 0.5 to 56 μM, the seven most potent and selective inhibitors exhibit mixed, mostly competitive inhibition. These molecules were included in the recently developed homology model of h-ENPP1. All exhibit competitive inhibition by endogenous ligands and adopt binding patterns akin to those of ATP. The selectivity of ENPP1 over ENPP3 can be explained by the electrostatic potential of the two proteins; ENPP1 prefers negative ligands. The inhibitor with the minimal Ki,app (0.5 μM) value (X = CH2, Y = BH3, W = O, R = H, n = 1) is also inactive against P2Y receptors. In general, derivatives with X = CH2, Y = BH3, W = O, R = H, and n = 1 are the most potent and selective ENPP1 inhibitors (Fig. 7) [54].

Nucleotide-based ENPP inhibitors
Non-nucleotide-based inhibitors
Biphenyl oxazole derivative–based inhibitors
H. Ahmed et al. synthesized biphenyl oxazole derivative 8 in excellent yield by using Suzuki-Miyaura cross-coupling of bromophenyloxazole with different boronic acids and evaluated against ENPP1 and ENNP3 at 100 μM cancentration for ENPP1 and ENPP3 activity. Among the synthetical substrate thymidine analogs, they found two compounds that are potent and specific inhibitors of both enzymes: compound 9 inhibits ENPP1 with an IC50 of 0.15 μM; 10 inhibits ENPP3 with IC50 of 0.17 μM (Fig. 8) [55].

Biphenyl oxazole–, sulfonate-, and sulfamate derivative–based inhibitors
Sulfonate- and sulfamate derivative–based inhibitors
Various benzofuran and benzothiophene sulfonate and sulfamate derivatives 11 have been developed as potent and specific inhibitors of ENPP1 and ENPP3 by Semreen and co. With IC50 values varying from 0.12 to 0.95 µM, compounds 11a, 11b, 11c, and 11d are the most effective inhibitors of ENPP1. Compounds 11e, 11f, 11g, and 11h were the most effective ENPP3 inhibitors, with IC50 values extending from 0.12 to 0.95 μM. Although compound 11 with substituents (R = n-Pr, NHMe, X = S) is more selective for ENPP3 than ENPP1, compound 11 with substituents (R = Cy, p-Tolyl, NMe2) also shows ENPP1 selectivity over ENPP3. MDS indicates that the drug inhibitor suramin has similar binding properties to this drug. In this form, the zinc ion of the active site lies next to the sulfonate group, which functions as a cation-binding moiety. MDS indicate that the inhibitor suramin has similar binding properties to these drugs. In this mode, the sulfonate group acts as a cation-binding moiety close to the zinc ion in the active site (Fig. 8) [56]. Ullah et al. designed raloxifene sulfonate 12 or sulfamate 13 derivatives. The inhibitory effects of the drug target on ENPP1 and ENPP3 enzymes were evaluated. With an IC50 of 1.4 μM, compound 12a exhibited the highest activity against HT-29 colon cancer cells, outperforming F180 fibroblast cells by an 8.43-fold margin. Compound 12a demonstrated submicromolar IC50 values (IC50 = 0.29 µM and 0.71 µM, correspondingly) in relation to ENPP1 and ENPP3. ENPP1 homology structure and ENPP3 crystal structure were combined with the best inhibitors. All docked derivatives showed negative interactions in the active pockets of ENPP1 and ENPP3 [57]. Patel et al. designed a group of sulfonate derivatives 14 which have been tested as inhibitors of ENPP. Most drugs have been found to be effective in neutralizing the inhibitory effects of the ENPP1, ENPP2, and ENPP3 isoenzymes. Compound 14a is a potent and specific inhibitor of ENPP1 with an IC50 of 0.387 ± 0.007 μM. However, the most potent ENPP3 inhibitor was found to be 14b with an IC50 value of 0.214 ± 0.012 µM. The most potent ENPP2 inhibitor compound 14c has an IC50 of 0.659 ± 0.007 µM [58]. Jung et al. reported the synthesis of arylamide sulfonate derivative 15 and tested for its ability to inhibit ENPP1 and ENPP3 isoenzymes. Among the chosen inhibitors of ENPP1, the sub-micromolar IC50 values of compounds 15a and 15b were 0.28 ± 0.08 and 0.37 ± 0.03μM, respectively, and the IC50 of 16a was 0.81 ± 0.05μM. Selective inhibitors of the isoenzyme ENPP3 are 15c and 16b, which tend to reduce the action to half the maximum inhibiting intensity, which is 0.15 ± 0.04 and 0.16 ± 0.01 μM, respectively. Additionally, 15d was a more potent compound with IC50 values of 0.45 ± 0.07 µM against ENPP1 and 0.19 ± 0.02 µM against ENPP3. Enzyme kinetic studies of compound 15e showed that it non-competitively inhibits the ENPP1 isoenzyme, while compound 16c competitively terminates the activity of ENPP3 (Fig. 8) [59].
Sulfamide derivative–based inhibitors
Quinazoline-4-piperidine-4-ethylsulfonamide derivatives 17 and 18 were synthesized and tested as ENPP1 inhibitors. Nevertheless, this series has an issue with its extreme correspondence attachment to hERG potassium channels, which might result in QT prolongation. It retains ENPP1 activity but does not bind to hERG to demonstrate the interaction of compound with hERG (Fig. 9) [60].

Sulfamide-, pyrimidine-, quinoline-, and sulfonylurea derivative–based inhibitors
Pyrimidine derivative–based inhibitor
Ausekle et al. designed dihydropyrimidopyrimidinone 19 and 3,4-dihydropyridopyrimidinone 20 analogs that have repressive actions on ENPP1. The development of 19 and 20a as strong ENPP1 inhibitors was prompted by SAR results. In addition, human, mouse, and rat liver microsomes showed strong microsomal stability of compounds 19 and 20a. Additionally, 19 and 20a did not inhibit CYP (1A2, 2C9, 2C19, 2D6, and 3A4). The binding mechanism of ENPP1 and drugs (19 and 20a) was understood by MDS experiments (Fig. 9) [61].
Quinoline derivative–based inhibitors
Based on chemo- and regioselective Suzuki processes, Ullah al. synthesized the substituted arylated trifluoromethylquinoline derivative 21. The produced compounds were demonstrated to be promising and selective h-ENPP inhibitors when associated to h-NTPDases. The majority of these substances were discovered to be weakly inhibiting h-ENPP3 and selectively inhibiting h-ENPP1. It was discovered that most of these substances had minimal h-ENPP3 inhibition and were very selective h-ENPP1 inhibitors. It was shown that only four chemicals effectively inhibited h-ENPP3: 21a (Br at positions 3 and 8), 21b (Br at positions 3, 4, and 8), 21c, and 21d (Br at position 3). Compound 21d had the highest level of efficacy in suppressing h-ENPP3, with an IC50 value of 0.36 ± 0.04 µM. With an IC50 value of 0.25 ± 0.02 µM, derivative 5d was the second most effective drug for suppressing h-ENPP1. Due to their unique therapeutic importance, these molecules will be further analyzed to see whether they can work as therapeutic targets (Fig. 9) [62]. Choudhary et al. produced various N-Fused isoquinoline derivatives 22. The 16 compounds produced were screened for their potential h-ENPP-1 and h-ENPP-3 inhibitory properties. Using synthetic p-Nph-5ʹ-TMP, the inhibitory activity of these drugs against h-ENPP-1 and h-ENPP-3 was evaluated. The findings indicated that 22b was a strong inhibitor of h-ENPP-3 (IC50 = 0.48 ± 0.01 µM), while compound 22a was the greatest inhibitor of h-ENPP-1 (IC50 = 0.36 ± 0.06 µM). MDS reveled that the both compounds have strong \(\pi -\pi\) stacking interaction with Tyr340 and \(\pi -\sigma\) interaction with His329 and one hydrogen bond interaction with Lys204 (Fig. 9) [63].
Sulfonylurea derivative–based inhibitors
Khan et al. reported the synthesis of sulfonylurea derivatives containing pyrrolopyridine core 23 as an inhibitor of the ENPP1 and ENPP3 isozymes that are over-expressed in cancer and diabetes. The compound 23c was determined to be the most efficient ENPP1 inhibitor, with an IC50 value of 0.80 ± 0.04 µM, whereas 23a was identified by enzyme analysis as a selective ENPP1 inhibitor. The most effective and moderately selective ENPP3 inhibitor was revealed to be derivative 23b (IC50 = 0.55 ± 0.01 µM) (Fig. 9) [64].
Biscoumarin derivative–based inhibitors
The inhibitory effect of dicoumarin derivative 24 on snake venom and ENPP1 enzyme was tested. Based on the secondary transformations and Lineweaver-Burk and Dixon plots, it can be concluded that these compounds are non-competitive inhibitors of both enzymes. It was determined that the Ki and IC50 values of biscoumarin for the human recombinant ENPP1 enzyme ranged from 50 to 1000 and 164 to > 1000 µM, respectively, while the Ki and IC50 values for snake venom phosphodiesterase ranged from 1150 to 9.44 and from 9.44 to > 1000 µM. Compounds 24a, 24b, 24c, 24d, 24e, 24f, 24g, and 24h were found to be non-competitive and non-cytotoxic at concentrations up to 200 µg/mL, with cell death below 10% after 3 h of incubation (Fig. 10) [65].

Biscoumarin-, oxadiazole-, thiadiazole-, triazole-, imidazole-, pyrrolopyrimidine-, pyrrolopyridine-, and pyrazolo-pyridinone-based inhibitors
Oxadiazole- and thiadiazole derivative–based inhibitors
The derivatives of 1,3,4-oxadiazole-2 (3H)-thione 25 and 1,3,4-thiadiazole-2 (3H)-thione 26 were synthesized and their inhibitory effects against two ENPP1 enzymes were examined. Because the Vmax value drops in the absence of intervention and Km is considerable, the Dixon and Lineweaver-Burk plots and their second transformations demonstrate that the inhibition of snake venom and pure human recombinase is not competitive. Based on their respective IC50 values of 368 µM and 66.47 µM, derivatives 26a and 25a were determined to be the most active molecule. For human recombinant ENPP1 enzymes and snake venom, the corresponding Ki values are 360 µM and 100 µM. It has been shown that most active drugs do not have toxicity in terms of neutrophil survival (Fig. 10) [66].
Triazole derivative–based inhibitors
A group of Schiff-based triazoles 27 was created and assessed for their capability to stop ENPP1. Out of the 25 compounds, three were well-known as powerful inhibitors with higher activity compared to conventional EDTA (IC50 = 277.69 ± 2.52 µM): 27a (IC50 = 132.20 ± 2.89 µM), 27b (IC50 = 152.83 ± 2.39 µM), and 27c (IC50 = 251.0 ± 6.64 µM) (Fig. 10) [67].
Imidazole derivative–based inhibitors
The discovery of thiazolo[3,2-a]benzimidazol-3(2H)-one analogues 28 as a novel inhibitor of ENPP1 which have drug-like features. 28a was revealed to be the most effective ENPP1 inhibitor out of the 25 compounds that were investigated in this investigation. When using ATP as a substrate, its Ki value is 467 nM, and its mechanism of non-competitive inhibition is present (Fig. 10) [68].
Pyrrolopyrimidine- and pyrrolopyridine derivative–based inhibitors
SAR research was carried out together with the design and synthesis of pyrrolopyrimidine 29 and pyrrolopyridine derivative 30. They discovered that 29a stimulates the STING pathway in a concentration-dependent manner and has a very strong (IC50 ± 25.0 µM) anti-ENPP1 effect. Furthermore, 29a produces cytokines (including IP-10 and IFN-β) in a concentration-dependent manner in response to STING activation. Ultimately, they discovered that in the 4T1 genetic mouse model, 29a suppressed tumor development. They offer fresh perspectives on the creation of novel ENPP1 inhibitors and lay the groundwork for the further advancement of tiny antibodies intended for use in cancer treatment (Fig. 10) [69].
Pyrazolo-pyridinone derivative–based inhibitors
Arif et al. cyclized electron-rich 3-amino-1H-pyrazoles with 1,3-diketones to generate fluorinated and non-fluorinated pyrazolopyridinone 31. The capability of these substances to suppress human recombinant ALP and ENPP enzymes was assessed. The findings of the in vitro bioassay demonstrated both target enzymes’ specific and strong inhibition. Compound 31a had the maximum degree of h-TNAP selectivity at the tested dosages, whereas compound 31b preferentially inhibited the h-ALP isoenzyme. Significantly, compounds 31c and 31d resemble human ENPP1 and ENPP3 lead scaffolds, respectively (Fig. 10) [70].
Thiadiazolopyrimidone derivative–based inhibitors
Gangar used the Suzuki-Miyaura reaction to create 2-aryl-1,3,4-thiadiazolopyrimidine and its 6-fluoro derivative 32. It has been determined that three substances are specific ENPP inhibitors. Out of all the derivatives, compound 32a had the most inhibitory potential for h-ENPP1 (IC50 ± SEM = 0.39 ± 0.01 µM), whereas compound 32b had the highest inhibitory potential for h-ENPP3 (IC50 ± SEM = 1.02 ± 0.05 µM). Derivative 32c (IC50 ± SEM = 0.31 ± 0.01 µM) demonstrated the most inhibitory action on ENPP1 among the fluorinated thiadiazolopyrimidinones, and it was found to be equivalent to controls like suramin (IC50 ± SEM = 8.67 ± 1 µM). Furthermore, MDS and homology modeling were run on both inhibitors to infer the inhibitors’ mechanism of binding with the corresponding enzymes (e.g., h-ENPP1 and h-ENPP3) (Fig. 11) [71].

Thiadiazolopyrimidone-, hydrazine-, thioguanine-, imidazopyridine-, and purine-thioacetamide-based inhibitors
Hydrazine derivative–based inhibitors
Chang et al. synthesized new diacylhydrazine derivatives 33 that are potential inhibitors of ENPPs. It was found that among different derivatives, compound (33) showed the greatest inhibition of the two isozymes. The best inhibitory activities were (34a) (IC50 ± SEM = 1.59 ± 0.25 μM) and (34b) (IC50 ± SEM = 1.07 ± 0.12 μM), which showed good and uncompetitive in the receptor region of the h-ENPP3 inhibitory mechanism compounds and h-ENPP1 respectively (Fig. 11) [72].
Thioguanine derivative–based inhibitors
A group of brand-new, non-nucleotide small-molecule ENPP1 inhibitors based on thioguanine 35 was synthesized. Lead chemical 35a demonstrated strong anti-inflammatory activities in vivo along with excellent in vitro potency, selectivity, stability in SGF/SIF/PBS, and ADME and pharmacokinetic parameters. The high microsomal stability investigation revealed that our lead chemical 35a (Ki = 41 µM) exhibited good effectiveness with no elimination (MLM: 12.9 μL/min/mg and HLM: 6.3). Additional in vitro ADME metrics for compound 35a include plasma stability (percentage of drug left in plasma after 5 h in humans and mice, 89.5% and 75.8%, respectively) and percent free drug in plasma at 5 h in rats and humans, 37% and 47.5%, respectively (Fig. 11) [73].
Imidazopyridine- and purine-thioacetamide derivative–based inhibitors
Bowman et al. reported ENPP inhibitor library that includes compounds like p-nitrophenyl 5′-thymidine monophosphate (p-Nph-5′-TMP) 36. Compound 36a, which was discovered by high-throughput analysis of a colorimetric experiment utilizing the substrate p-Nph-5'-TMP, is a new and effective inhibitor of human ENPP1 (Ki = 0.217 μM). Different bicyclic scaffolds (imidazo[4,5-b]pyridine, imidazo[4,5-c]pyridine, imidazo[4,5-b]pyrazine, and purine) are catalyzed by ENPP1, according to SAR. Purine and imidazole-[4,5-b]pyridine, two of the primary samples, were chosen for further SAR analysis. At all dosages, strong inhibitors of ENPP1 were detected, with Ki values of 29.6 nM for imidazo[4,5-b]pyridine 36c and 5.00 nM for purine analog 36b. Compound 36c, the most powerful ATP-hydrolyzing inhibitor of ENPP1, has a selectivity of 13 times greater than that of the extracellular ENPP isozymes ENPP2 and ENPP3 (Fig. 11) [74].
Inhibitor of ecto-5′-nucleotidase (CD73)
Nucleotide-based inhibitors
Three new cytosine-derived α,β-methylene diphosphonates, 37, 37a, and 37b, were assessed for their capability to stop membrane-bound CD73 activity in primary astrocytes in vitro within the concentration range of 1 × 10−9 to 1 × 10−3 M. Every investigated chemical has a low nanomolar range Ki value with good binding capacity and a maximal inhibition of around 1 × 10−3 M with submicromolar range IC50 value. Derivative 37 among all had IC50 and Ki values of 18.2 nM and 0.11 μM, respectively. Even though it was tested at a concentration much above its IC50 value, derivative 37 was the only substance that could cause the CD73 to shed from astrocyte membranes and improve astrocyte movement in the scratch wound passage test [75]. A phase I clinical trial is starting to evaluate AB680 38, a potent inhibitor of human CD73, for its effectiveness in treating solid tumors. To identify the mechanism of inhibition, they performed a thorough kinetic investigation of the relations between human CD73 and compound 38. Compound 38 was discovered to be a slow-onset, reversible competitive inhibitor of human CD73, having a Ki of 5 pM [76]. The compound 38, a strong (Ki = 5 pM), reversible, and selective inhibitor of CD73, has prompted extensive research in SAR, drug-based model building, and pharmacokinetic optimization. Additionally, compound 38 has an extended half-life and minimal clearance in preclinical strains, which contributes to a PK profile that is advantageous for the parenteral processing’s long-term action [77]. A class of monophosphate small-molecule CD73 inhibitors was created. Together with, OP-5244 (39) has been proven to be an effective and highly bioavailable CD73 inhibitor with a biochemical IC50 value in the range of 0.25 ± 0.08 nM. Preclinical research revealed that compound 39 totally stopped human cancer cells and CD8+ T lymphocytes from producing ADO. Furthermore, compound 39 reversed the immunological response and decreased the ADO/AMP ratio in mouse models, suggesting its promise as an in vivo means for future research [78]. Junker et al. produced a family of CD73 inhibitors based on methylenephosphonic acid 40 by employing structure-based design. SAR studies performed on this model showed that 40a has an IC50 of 2.6 nM, shows good activity against CD73, is highly selective for exonucleases, and has good pharmacokinetic properties (Fig. 12) [79].

Nucleotide-based ecto-5′-nucleotidase inhibitors
Bhattarai et al. developed a series of adenosine-5′-methylphosphonic acid derivatives 41. The derived nucleotides 41 underwent substitutions at the side chain’s methylene group or modifications at the N6-, C8-, and both locations of the adenine moiety of 41. The produced nucleotides were assessed for their ability to block CD73. All chemicals have Ki in the minimal nanomolar range, according to SAR. Efficacy was improved by replacement of N6-benzyl, N6-(2-phenylethyl), N6-(4-chlorobenzyl), and O6-benzyl. 41a, 41b, and 41c were the most potent inhibitors, with Ki values of 7.23 nM, 8.04 nM, and 9.03 nM, respectively. Equally strong inhibitors were produced when the 6-NH group was replaced by O (41d) or S (41e), both of which are analogs of 41c (41d, Ki = 9.20 nM; 41e, Ki = 9.50 nM) [80]. Bhattarai et al. synthesized a set of 50 nucleoside 5′-α,β-methylene-diphosphates 42–44 based on purines and pyrimidines, which were CD73 inhibitors. All chemicals have a nanomolar range Ki value, according to SAR. 42a, 43a, 44a, and 44b were the most effective inhibitors at rat CD73, with Ki = 14.8 nM, 13.9 nM, 18.8 nM, and 3.67 nM, respectively. It has been demonstrated that compound 44b is more selective for CD73 than cytosolic 5′-nucleotidase and UDP-activated P2Y (P2Y6 and P2Y14) receptors. (Fig. 12) [81].
R. Ghoteimi et al. studied SAR of novel derivatives of α,β-methylene-ADP (AOPCP)45 substituted in the 2-position as CD73 inhibitors. With Ki values on human CD73 of 3–6 nM, they discovered that the most prevalent potent inhibitors are 2-iodo and 2-chloro derivatives (45a, 45b). By using X-ray crystallography, different binding modes were found, depending on the size and type of the 2-substituent. Depending on the kind of attachment, species variations were noted. For instance, 2-piperazinyl-AOPCP (45c) exhibited a more than 12-fold reduction in its ability to bind to mouse CD73 in contrast to human CD73. This work demonstrated that adding the big N6 product was not necessary to obtain strong CD73 inhibitory potential; instead, a minor mutation at position 2 of AOPCP may be introduced [82]. Liu et al. built and acquired the X-ray cocrystal structure of human CD73 complexed with nucleotide analog 46 as an inhibitor. The novel CD73 inhibitor 46a shows excellent potency, selectivity, and metabolic stability with subnanomolar Ki values of 0.316 ± 0.020 nM and 0.746 ± 0.246 nM in humans and mice, respectively. They found that compound 46a is the most potent inhibitor of CD73 for recombinant CD73 and native CD73 which are present in cancer as well as epithelial cell in mouse and human tissue. The most important thing is that for 46a, there is no risk of formation of adenosine receptor–activating compounds, which lead to serious side effects [83]. A number of substituted 5′-aminonucleotide analogs 47 were created. Phosphonic acids 47a and derivatives 47b and 47c with the purified recombinant protein demonstrated marginal suppression of CD73 in the cell-based test (45–61% inhibition at 100 µM and 46–52% inhibition at 100 µM, respectively). They speculate that the reason for this discrepancy might be because soluble protein forms were used in the experiments, whereas the protein’s membrane fixation in cell-based tests could be the cause. Derivatives can reach and/or adapt to the protein’s catalytic site in different ways in both situations. [84]. The synthesis of new CD73 inhibitors by the replacement of bis-phosphonic acid with methylenephosphonic acid 48 which increases the stability of the compound. Clinical evolution shows that combination with monoclonal antibodies targeting the immune system is extremely predicted [85]. A series of CD73 inhibitors were developed through molecular docking, 3D-QSAR 49, and studied to reveal their SAR. Relations among inhibitors and protein are studied through MDS. Later, CoMFA and CoMSIA developed a 3D-QSAR model. The optimal CoMSIA model has Q2 and R2 values of 0.809 and 0.992, respectively, whereas the optimal CoMFA model has Q2 and R2 values of 0.708 and 0.983, respectively. Furthermore, MDS was used to assess the stability of the complex produced by the two inhibitors and CD73; the outcomes were in line with those of investigations using molecular docking and 3D-QSAR [86]. Analogs of nucleotides 50 were created by substituting an aromatic ring or a purine residue with a triazole moiety, and they were then assessed for their ability to inhibit CD73. Adenosine-mediated immunosuppression of human T cells was reversed by the most potent inhibitors, 50a and 50b, which contained bis(trifluoromethyl)phenyl or naphthyl substituents and showed IC50 values of 4.8 ± 0.8 µM and 0.86 ± 0.2 µM, respectively, in comparison to the standard AOPCP (IC50 value of 3.8 ± 0.9 µM) (Fig. 13) [87].

Nucleotide-based ecto-5′-nucleotidase inhibitors
Non-nucleotide-based inhibitors
Thioxoimidazolidinone derivative–based inhibitors
Derivatives of azomethine–thioxoimidazolidin 51 was tested for enzyme inhibition using an isozyme that is both human and rat. 51a exhibited significant inhibition against h-CD73, with an IC50 value of 0.23 ± 0.08 µM, while, two other substances, 51b and 51c, had strong inhibitory activity that was not selective against rat and human enzymes. Furthermore, these compounds (51a, 51b, and 51c) were further investigated for their impact on the quantifiable real-time polymerase chain reaction demonstration of h-CD73 (Fig. 14) [88].

Thioxoimidazolidinone, triazole and thiazole, sulfonic acids, phelligridin, and hydroxamic acid–derived CD73 inhibitors
Triazole- and thiazole derivative–based inhibitors
An aromatic ring including moiety 52 and 53 that are 1,4-disubstituted 1,2,3-triazoles was created, and its potential to inhibit CD73 expression was assessed. The compounds 52a, 52b, 53a, 53b, and 53c had the highest potency at 10 μM, whereas over 20 derivatives demonstrated greater inhibition at 80% of hCD73 at 100 μM. However, compared to the original RR3, these medications are weaker inhibitors. A poor activity can result from a variety of variables, including the conversion of the imidazole scaffold to the triazole scaffold and the type and length of the linker, which can influence how attractive the target protein is for contact [89]. A new class of benzotriazole derivative 54 was introduced as inhibitors of CD73. They found that the most potent inhibitors were 54a with an IC50 = 12 nM and 54b showing an IC50 = 19 nM. The competitive binding mechanism of 54b was found during cocrystallization with human CD73. Because these compounds lessen the limited membrane permeability and basic acidity of established CD73 nucleoside inhibitors, they should improve drug-like characteristics [90]. A thiazole derivative 55 was synthesized and evaluated the ability to inhibit CD73 against both human and rat CD73. The derivative 55a was showing maximum inhibition against h-CD73 with IC50 value 0.32 ± 0.03 µM. This value is 24-fold greater than its action towards r-CD73. Additionally, molecular docking was performed to identify relevant binding sites (Fig. 14) [91].
Sulfonic acid derivative–based inhibitors
Sulfonic acid derivative 56 was identified as a potent inhibitor of CD73. The most valuable potent inhibitor was revealed to be 56a, which replaced naphthalene for sulfonic acid. The rat enzyme’s IC50 value was 10.4 ± 3.3 µM, whereas the human enzymes were 1.32 ± 0.09 µM. All substances are generally more active against human enzymes. SAR was created for this novel inhibitor family. On the H157 cancer cell line, several sulfonic acid inhibitors have also been shown to be strong cytotoxic medications [92]. The investigation of biochemical properties of human and rat CD73 than characterization of sulfonic acid derivatives elaborated that it acts as potential inhibitors of CD73. The highest number of potent inhibitors for rat and human CD73 was 57 and 58, with a Ki value of 0.78 µM and 0.66 µM, respectively (Fig. 14) [93].
Phelligridin derivative–based inhibitors
An enzyme-based test and computer-aided drug discovery were used to identify a new CD73 inhibitor. A total of 500 compounds with an elevated binding similarity were extracted from the Chemdiv-Plus database via structure-based virtual screening. At a concentration of 100 μM, the compounds’ inhibitory value against CD73 enzyme activity was assessed. Twenty compounds exhibited an inhibitory value more than 20%; eight of these chemicals had dose dependent IC50 values varying from 6.72 to 172.1 μM. With an IC50 value of 6.72 µM and an inhibitory activity within the range of 95.52 ± 0.12%, compound 59 was determined to be the most effective potent inhibitor. Phelligridin-based compounds have the best experimental inhibitory values among the studied substances (Fig. 14) [94].
Hydroxamic acid derivative–based inhibitors
The study of hydroxamic acid-containing compounds as potential human CD73 inhibitors, because this group is known to be a strong chelator of zinc. Twelve of the 25 derivatives that were considered were validated experimentally by VS and then put through enzymatic analysis. It was discovered that four of these (33.3%) inhibited h-CD73 at small micromolar concentrations. 6.2 ± 1.0 μM was the IC50 value of the most powerful. All inhibitors met the requirements for a drug-like structure and offered novel scaffolds that may be investigated in later stages for additional optimization (Fig. 14) [95].
Spirooxindole derivative–based inhibitors
A spirooxindole derivative 61 was identified as a potent inhibitor of human as well as rat CD73 enzymes. They found that the most potent inhibitor was compound 61a which showed 280-fold higher inhibition with an IC50 = 0.15 ± 0.02 μM and compound 61b (IC50 ± 0.19 ± 0.03 μM) on CD73 with 406-fold greater inhibition than reference standard sulfamic acid (Fig. 15) [96].

Spirooxindole-, quinoline-, coumarin-, sulfonamide-, anthraquinone-, and pyridine-based inhibitors
Quinoline derivative–based inhibitors
A method for the synthesis of diarylated quinoline 62 including two identical aryl groups using Suzuki-Miyaura cross-coupling was developed and evaluated as potential inhibitors of the rat and h-CD73 isozyme. Most of the derivatives showed selective inhibition of h-CD73 with considerable IC50 values. The most potent inhibitors are 62a, 62b, 62c, and 62d and all have IC50 values > 100 µM (Fig. 15) [97].
Coumarin derivative–based inhibitors
A heteroannulated pyrido[2,3-c]-coumarin 63 was synthesized by using domino reactions and found a novel inhibitor of CD73. Compound 63a was showed strong inhibition against h-CD73 as well as r-CD73 with an IC50 value in the range of 3.95 ± 0.12 and 2.67 ± 0.03 µM, respectively (Fig. 15) [98].
Sulfonamide derivative–based inhibitors
Miliutina et al. test 51 compounds as inhibitors of CD73 and found that only 13 were capable of inhibiting CD73. Out of 51 potential compounds, the most potent inhibitors were chosen for experimental assessment. It was determined that 13 of these compounds exhibited competitive inhibitory action. Sulfamoylphenyl-2H-chromene-3-carboxylic acid amide (64), with an IC50 value of 1.90 μM, was the most effective inhibitor, while other nucleotide and anthraquinone-based compounds have drug-like structure but different from structure of known active compounds (Fig. 15) [99].
Anthraquinone derivative–based inhibitors
The investigation of inhibitory activities of different anthraquinone derivative 65 against CD73 showed that only few derivatives have Ki value in low micromolar range between 1 and 7 μM, while five exhibited even sub-micromolar Ki values between 0.15 and 0.6 μM. The derivatives 65a with Ki value 260 nM and 65b with Ki value 150 nM are the most potent inhibitors of CD73. P2Y receptor subtypes P2Y2, P2Y4, P2Y6, and P2Y12, as well as NTPDases, were studied. It was shown that compound 65a had the highest selectivity (> 150-fold) (Fig. 15) [100].
Pyridine derivative–based inhibitors
Zhang et al. reported the synthesis of pyrazolo[3,4-b]pyridines, pyrrolo[2,3-b] pyridines, pyrido[2,3-d]pyrimidines 66, and benzofuro[3,2-b]pyridines 67. These compounds are very attractive because they have good fluorescent properties and significant abilities to inhibit CD73 and potentially induce cytotoxic activity. Among the tested compounds, 66a was found to be a selective inhibitor for human CD73 exhibiting IC50 value in human is 0.32 ± 0.05 µM, while 66b was an inhibitor of mouse CD73 showing that IC50 value is 0.67 ± 0.12 µM (Fig. 15) [101].
Inhibitor of ectonucleoside triphosphate diphosphohydrolases (NTPDases)
There are many compounds are reported as inhibitor of NTPDase. There are two main categories of inhibitors of NTPDases: nucleotide-based inhibitors and non-nucleotide-based inhibitors. While ARL67156 68, 8-BuS-ATP 69, and PSB-6426 70 are nucleotide-based inhibitors, non-nucleotide-based inhibitors refer to compounds that do not contain a nucleotide structure. PPADS 71, suramin 72, tryptamine-derived imine 73, reactive blue-2 74, and its derivative PSB-071 75 are examples of non-nucleotide inhibitors (Fig. 16) [102,103,104].

Some standard NTPDase inhibitors
Nucleotide-based inhibitors
Gendron et al. investigated that ARL 67156 (76) is a weak inhibitor of NTPDase1, 3 and ENPP1, but not an effective inhibitor of NTPDase2, ENPP3, and CD73. First, our results show that at the concentrations most commonly used in the cellular environment (50–100 µM), 76 ATP binding to P2 receptors will be long-acting if NTPDase1, NTPDase3, or ENPP1 are the main ectonucleotidases in the study. Second, our biochemical data indicate that 76 will not inhibit ATP hydrolysis in assays using high concentrations of ectonucleotidase nucleotides or in cells expressing NTPDase2 or ENPP3. Therefore, some precautions need to be taken when using 76 [105]. Gillerman et al. reported that 8-BuS-AMP (77) and 8-BuS-ADP (77a) analogs can be used to induce or inhibit NTPDase1 activity for various purposes in vitro and potentially in vivo. For human NTPDase1, the Ki values of analogs 77 and 77a were assessed to be 0.8 and 0.9 mM, respectively. These novel inhibitors also open therapeutic avenues for platelet homeostasis, immunity, and cancer, specifically by blocking NTPDase1 [106]. ATP analogs were developed as inhibitors of NTPDase 78. Among the synthesized analogs,8-BuS-ATP 78a was created to be the best non-hydrolyzable competitive inhibitor with a Ki value of approximately 10 μM. This non-hydrolyzable analog did not antagonize P2X receptor–mediated effects on non-endothelial unlined blood vessels in guinea pig mesenteric beds [107]. The derivatives of tri- and monophosphate 79 were synthesized and evaluated as NTPDase inhibitors. They found that the most selective and potent inhibitor of NTPDase2 among all compounds was 79a. Compound 79a is stable against hydrolysis by NTPDase1, 2, 3, and 8. It inhibits h-NTPDase2 with a Ki of 20 μM and only small amounts (5–15%) of NTPDase1, 3, and 8. Homology molds of h-NTPDase1 and 2 were composed. The selectivity of 79a for NTPDase2 over NTPDase1 is due to the thiohexyl portion of 79a being well-positioned in the hydrophobic pocket; however, in NTPDase1, it is revealed to the solvent [108]. Adenine and uracil nucleotide derivative 80 was synthesized by replacing phosphate group with phosphonic acid ester at 5ʹ position of nucleotide by amide linker. The most potent compound is 80a, which is a competitive inhibitor of NTPDase2 and presents a Ki value of 8.2 µM and selectivity compared to other NTPDases. It is inactive against uracil nucleotide–activated P2Y2, P2Y4, and P2Y6 receptor subtypes. Derivative 80a is chemically and metabolically stable. Unlike many known (non-selective) NTPDase inhibitors, 80a is uncharged and has oral bioavailability (Fig. 17) [109].

Nucleotide-based NTPDase inhibitors
Non-nucleotide-based inhibitors
Oxoindolin hydrazine carboxamide derivative–based inhibitors
Oxoindolin phenylhydrazine carboxamide derivatives 81 were synthesized and were evaluated as potent inhibitor of h-NTPDase. The most potent inhibiters were compounds 81a with IC50 value of 0.12 ± 0.03 µM for NTPDase1, 81b with IC50 0.15 ± 0.01 µM for h-NTPDase2, and 81c with IC50 value of 0.30 ± 0.04 for hNTPDase3 and 0.16 ± 0.02 µM for h-NTPDase8. Four compounds (81d, 81e, 81f, and 81a) were linked with the discriminatory inhibition of h-NTPDase1 while 81g was recognized as a selective h-NTPDase3 inhibitor. Additionally, the utmost effective inhibitors were docked within the active site of enzyme and the monitored interfaces were in accordance with in vitro results [110]. The other approach is in the synthesis of oxoindolin hydrazine carbothioamide derivatives 82 and evaluated as inhibitors of NTPDase. The most potent inhibitors were found to be 82a, 82b, 82c, 82d, and 82e for NTPDase1 with an IC50 value 0.29 ± 0.02 µM, 0.15 ± 0.009 µM, 0.24 ± 0.01 µM, 0.30 ± 0.03 µM, and 0.16 ± 0.01 µM respectively. Likewise, derivative 82e with IC50 0.16 ± 0.01 µM was noticed to be a selective h-NTPDase2 inhibitor. The most potent inhibitor for h-NTPDase3 was found to be derivatives 82f with IC50 0.19 ± 0.02 µM and 82g with IC50 0.38 ± 0.03 µM. MDS were also performed on the most active compounds to identify interaction sites. Therefore, this drug is an important tool to examine the function of NTPDase3 in insulin secretion (Fig. 18) [111].

Oxoindolin hydrazine carboxamide–, anthraquinone-, imidazothiazole-, or imidazooxazole-based inhibitors
Anthraquinone derivative–based inhibitors
The structure of anthraquinone derivatives 83 is related to anthraquinone dye reactive blue 2. The anthraquinone derivative was made and assessed as potent inhibitors of NTPDase. From all synthesized compounds, the most potent inhibitor was found to be 83a with an IC50 value of 539 nM and 83b with an IC50 value of 551 nM. The compounds 83c and 83d were found to be potent inhibitors of NTPDase3 with an IC50 value of 390 nM and 723 nM respectively [112]. The first crystal structures of an NTPDase catalytic ectodomain in association with the inhibitor PSB-071 (84), which is generated from RB-2, are shown by Zebisch et al. who studied that the inhibitor attaches to the nucleoside binding site as a sandwich formed of two molecules in each of the two crystal forms that have been shown. The orientation of one of the molecules is clearly defined. The nucleoside binding loop and the sulfonyl group form hydrogen bonds. Between R245 and R394, the latter of which is only present in NTPDase2, is the methylphenyl side chain functionality that increased NTPDase2-specificity. Since the second molecule has a lot of rotational mobility in-plane, it cannot be modeled in a certain orientation [113]. The effectiveness of 25 anthraquinone derivatives 85 linked to RB-2 at inhibiting rat NTPDases1, 2, and 3 was reported. NTPDases were inhibited by several 1-amino-2-sulfo-4-ar(alk)ylaminoanthraquinone derivatives in a concentration-dependent way. Given that the 2-methyl-substituted derivatives lacked inhibitory effect, it was discovered that the 2-sulfonate group was necessary for this activity. A non-selective competitive blocker of NTPDases1, 2, and 3 (Ki 16–18 μM) was found for 85a, while on the other hand, a powerful inhibitor with a predilection for NTPDase1 (Ki 0.328 μM) and NTPDase3 (Ki 2.22 μM) was found for 85b. A potent and specific inhibitor of rat NTPDase3, 85c was its isomer (Ki 1.5 μM) (Fig. 18) [114].
Imidazothiazole or imidazooxazole derivative–based inhibitors
Imidazothiazole- and imidazooxazole-based sulfonates and sulfamates 86 were synthesized and evaluated as inhibitors of all four isozyme NTPDases. The SAR analysis of these derivatives has shown fluctuations in their inhibitory strengths against distinct isoenzymes. Specifically, substituting the oxygen of the imidazooxazole core for the sulfur atom of the imidazothiazole core resulted in an enhanced sensitivity to NTPDase2. The findings indicated that some derivatives, such as compounds 86a, 86b, 86c, and 86d, were significantly more effective than the standard and showed more robust action against certain NTPDase isozymes than suramin. With an IC50 value of 0.36 µM, analog 86a is a discriminatory inhibitor of NTPDase1. With an IC50 value of 0.29 µM, analog 86b is a discerning inhibitor of NTPDase2. With an IC50 value of 0.37 µM, analog 86c is a discerning inhibitor of NTPDase3. Compound 86d, with an IC50 value of 0.36 µM, is the last selective inhibitor of NTPDase8. For the most promising drugs, molecular docking investigations were conducted against their specifically inhibited isoenzyme (Fig. 18) [115].
Quinoline derivative–based inhibitors
A series of substituted quinoline compounds, 87 and 88 was synthesized to assess them as NTPDase inhibitors The IC50 (µM) values of these quinoline derivatives ranged from 0.20 to 1.75, 0.77 to 2.20, 0.36 to 5.50, and 0.90 to 1.82 for NTPDase1, 2, 3, and 8, respectively. The most potent molecule against NTPDase1 was derivative 88a, which exhibited selectivity towards NTPDase1 and an IC50 of 0.20 ± 0.02 µM. Corresponding to NTPDase2, NTPDase3, and NTPdase8, derivatives 88b (IC50, 0.77 ± 0.06), 87a (IC50, 0.36 ± 0.01), and 88b (IC50, 0.90 ± 0.08) showed good activity [116]. The highly functionalized 2-arylquinoline derivatives 89 were synthesized and assessed as hNTPDase1, 2, 3, and 8 inhibitors. Inhibiting hNTPDase1 and/or h-NTPDase8 was possible for most substances. Two substances, 89a and 89b, were illustrated to be selective inhibitors of h-NTPDase1, with IC50 values of 13.9 ± 0.06 and 29.3 ± 0.72 µM, in that order. Compound 89c, on the other hand, specifically inhibited h-NTPDase8 (IC50 value 8.99 ± 0.67µM). The most effective inhibitors of hNTPDase1, 2, 3, and 8 were discovered to be the substances 89d (IC50 = 0.23 ± 0.01 µM), 89e (IC50 = 21.0 ± 0.03 µM), 89f (IC50 = 5.38 ± 0.21 µM), and 89g (IC50 = 1.13 ± 0.04 µM), in that order (Fig. 19) [117].

Quinoline-, pyrimidine-, thiadiazol-, polyoxotungstate-, tryptamine-, ticlopidine-, and triazinoindole-based inhibitors
Thiadiazole derivative–based inhibitors
Many thiadiazole amides (90) were created and assessed as strong NTPDase inhibitors. Most of the compounds were found to have encouraging inhibitory efficacy against h-NTPDase1, 2, and 8. Compounds 90a (0.05 ± 0.008 µM), 90b (0.04 ± 0.006 µM), and 90c (0.05 ± 0.01 µM) show remarkable inhibitory potential against h-NTPDase1, 2, and 8, in relation to their IC50 values. Nevertheless, only three substances—90d, 90c, and 90e—were able to inhibit h-NTPDase3. The h-NTPDase3 inhibitor with the highest efficacy among them was 90c, whose IC50 (0.38 ± 0.02 µM) was likewise less than 1 µM (Fig. 19) [118].
Pyrimidine derivative–based inhibitors
The thienopyrimidine derivatives (91) substituted with aryl and glycine were synthesized and assessed for their hydrolytic activity against the all four membrane-bounded isozymes of h-NTPDase. At 100 µM concentrations, we discovered very effective and specific inhibitors of every isozyme, with IC50 values varying from submicromolar to micromolar. Compound 91a (IC50 = 0.11 ± 0.03 µM) was evaluated as a selective structure against h-NTPDase1; compounds 91e (IC50 = 26 ± 2 µM), 91b (IC50 = 6 ± 0.04 µM), 91c (IC50 = 0.4 ± 0.03 µM), and 91g (IC50 = 0.13 ± 0.05 µM) were found to inhibit the activity of h-NTPDase2; compound 91d (IC50 = 3 ± 0.1 µM) was found to be a selective molecule against h-NTPDase3, but compound 91f (IC50 = 0.6 ± 0.5 µM) was found to be non-selective but the most active candidate against h-NTPDase8 (Fig. 19) [119].
Polyoxotungstate-based inhibitors
According to Müller et al., polyoxotungstates were shown to be powerful NTPDase1, 2, and 3 inhibitors. K6H2[TiW11CoO40], 92 was found to be the most powerful molecule showing Ki values of 0.140 µM for NTPdase1, 0.910 µM for NTPdase2, and 0.563 µM for NTPDase3. (NH4)18[NaSb9W21O86], 93 was one of the molecules that was specific to NTPDases2 and 3 as opposed to NTPDase1. NTPDase inhibition has been connected to the living impacts of polyoxometalates, especially their anti-cancer impact (Fig. 19) [120].
Tryptamine derivative–based inhibitors
Tryptamine Schiff bases 94 were synthesized and evaluated as NTPDase inhibitors. A total of 18 substances in all showed significant reduction of NTPDase1 (Ki = 0.0200–0.350 μM), 12 of NTPDase3 (Ki = 0.071–1.060 μM), and 15 of NTPDase8 (Ki = 0.0700–4.03 μM) activity. In contrast, the conventional inhibitor suramin’s Ki values were 1.260 ± 0.007, 6.39 ± 0.89, and 1.180 ± 0.002 μM, in that order. Lineweaver-Burk plot analysis revealed lead compounds (94a, 94b, and 94c) where all competitive inhibitors after kinetic investigations were conducted using human h-NTPDase1, 3, and 8 (Fig. 19) [121].
Ticlopidine derivative–based inhibitors
Previous research revealed that the recombinant version of human NTPDase1 (Ki = 14 μM) was inhibited by ticlopidine 95 in its prodrug form, which has no effect on P2 receptor activation. In this instance, they investigated ticlopidine’s potential as an NTPDase1 selective inhibitor. In several tests and forms, it was confirmed that ticlopidine 95 inhibits NTPDase1. The ADPase activity of COS-7 cells transfected with human NTPDase1 and intact HUVEC was considerably decreased by 100 µM ticlopidine, with reductions of 86% and 99%, respectively. NTPDase1’s ATPase activity in situ was completely inhibited by ticlopidine (100 µM), according to enzyme immunohistochemistry on human liver and pancreatic slices. Moreover, ticlopidine suppressed the activity of potato apyrase as well as rat and mouse NTPDase1. Ticlopidine at 100 µM had no effect on the endeavor of h-NTPDase2, 3, and 8; ENPP1; and ENPP3. NTPDase3 and 8 exhibited modest inhibition (10–20%) at 1 mM ticlopidine [122]. The other ticlopidine derivative 96 was produced, which was then tested for its ability to suppress human CD39. More powerful new CD39 inhibitors, 96a, 96b, 96c, and 96d, were obtained from the ticlopidine scaffold. On the other hand, thienotetrahydropyridines could function as prodrugs of P2Y12 receptor antagonists, which are triggered by the liver’s cytochrome P450 enzymes and cause an irreversible suppression of blood platelet aggregation. They thus used a benzene ring in place of the thiophene. Based on both scaffolds, several changes were made. A wide substitution has been made for the benzyl residue. With an IC50 value of 39.0 µM at CD39, inhibitor 96b is now the best option for additional research and development (Fig. 19) [123].
Triazinoindole derivative–based inhibitors
Based on triazinoindole 97, the designated compound acts as a CD39 inhibitor. The enzymatic activity of CD39 could be greatly suppressed by the identified inhibitor as well as one of its analogs, with IC50 values of 27.42 ± 5.52 and 79.24 ± 12.21 μM, respectively. Mutagenesis, microscale thermophoresis, and molecular docking studies suggested that residues like R85 could play a key function in the binding of triazinoindole molecules. The binding method may be employed for hit-to-lead optimization, and the recognized inhibitor can be more explored for its anti-cancer impact in vivo or utilized as a chemical agent to study CD39-related activities (Fig. 19) [124].
Inhibitor of alkaline phosphatases (ALPs)
Non-nucleotide-based inhibitors
Pyrazole-derived inhibitors
Pyrazole amide derivatives 98, which are strong TNAP inhibitors, were produced. Compound 98a is almost 200 times more potent against TNAP and shows high selectivity against the related PLAP isozyme, with an IC50 value in µM, after the hit-to-lead optimization of screening hit 1. Mechanistic studies revealed a distinct MOA for 98a, and in silico docking analyses supported these results [125]. A series of unique triazolyl pyrazole derivatives (99) were generated and evaluated as strong and selective inhibitors of h-TNAP over h-ENPP1 by incorporating a thiol carrying triazole moiety as the zinc binding functional group to a pyrazole-based pharmacophore. Numerous synthesized compounds were shown to be selective TNAP inhibitors by biological screening against h-TNAP, h-IALP, h-ENPP1, and h-ENPP3. Most of the compounds showed perfect selectivity and strong efficacy towards hTNAP in comparison to h-ENPP1. Compound 99a showed an extremely potent inhibitory action on hTNAP (IC50 = 0.16 µM or 160 nM), exhibiting a 127-fold increase in inhibition over levamisole. On the other hand, it was shown that compound 99b (IC50 = 1.59 ± 0.36 µM) was the most specific inhibitor against the studied ENPPs and ALPs. The explanation of selectivity between h-TNAP and h-IALP ligands and towards h-TNAP over h-ENPP1 was provided by binding site architectural exploration, molecular-docking, and MDS [126]. The aryl thiourea derivatives 100 of the non-steroidal anti-inflammatory medicine 4-aminophenazone, which is based on pyrazoles, were synthesized, as possible IALP enzyme inhibitors. When compound 100 is screened against IALP, lead member 100a is created; its IC50 value is 0.420 ± 0.012 µM, which is much better than the reference standard (l-phenylalanine IC50 = 100 ± 3.1 µM and KH2PO4 IC50 = 2.8 ± 0.06 µM). Based on kinetic studies that showed a non-competitive binding mode, SAR is utilized to recognize the binding pocket interaction of the active site and the way of enzyme inhibition. MDS was utilized to enhance the enzyme inhibition investigations by forecasting the protein behavior in response to active inhibitors 100a and 100b during the docking analysis (Fig. 20) [127].

Pyrazole-, pyrazolo-oxothiazolidine-, thiazole-, and thiophene-derived ALP inhibitors
Pyrazolo-oxothiazolidine-based inhibitors
The pyrazolo-oxothiazolidine compounds 101 were synthesized, and their in vitro effectiveness as ALP inhibitors was assessed. When compared to the standard reference KH2PO4 (IC50 = 5.242 ± 0.472 µM), 101a (IC50 = 0.045 ± 0.004 µM) showed the best inhibitory activity against ALP among all the synthesized compounds. To ascertain the ligands’ binding affinity with the target protein within the active site, molecular docking experiments were carried out. With ALP, all of the chemicals showed great docking binding affinities and high docking scores (Fig. 20) [128].
Thiazole- and thiophene derivative–based inhibitors
Several thiopheno-imidazo[2,1-b]thiazole derivatives 102 were synthesized and subsequently assessed for their potential as TNAP inhibitors. Using the same chemical motif, 102 a single chemical alteration results in a wide range of IC50 (from 42 ± 13 µM to more than 800 µM). The racemic thiophenyl derivative of levamisole (6HCl) 102a, as measured by porcine kidney TNAP, has an apparent IC50 of 42 ± 13 µM (n = 3, pH 10.4), which is twice as strong as the enantiomeric levamisole (IC50 = 93 ± 23 µM). This implies that enantiomeric thiophenyl compounds might be produced and refined for use in medicine to address pathological calcifications [129]. Compared to porcine kidney TNAP, the synthetic benzothiophene derivatives 103 and 104 showed more marked inhibitory characteristics towards BIAP. Using porcine kidney TNAP as a basis, two water-soluble racemic benzothiopheno-tetramisole and -2,3-dehydrotetramisole (103 HCl and 104 HCl) were identified, showing some potential for synthesizing and optimizing enantiomeric benzothiopheno-tetramisole, with apparent inhibition constants Ki = 85 ± 6 µM and 135 ± 3 µM comparable to that of enantiomeric levamisole, 93 ± 4 µM [130]. A series of benzocoumarin-thiazoles-azomethines (105) was created and examined their effects on human IALP and TNAP. While 105b (IC50 = 1.02 ± 0.04 μM) was suggested to be a potential inhibitor of h-IALP, 105a was determined to be the most effective h-TNAP inhibitor (IC50 = 0.76 ± 0.02 μM). In the human TNAP and IALP active site, the binding interactions were notable in comparison to the interactions demonstrated by reference standards (Fig. 20) [131].
Oxathiol-2-ylidene derivative–based inhibitors
The compound 1,3-oxathiol-2-ylidene benzamide (106) was synthesized and assessed for their capability to stop ALP. Nearly all the compounds exhibited excellent percentage inhibition against both enzymes, according to the data. While compounds 106b and 106c were revealed to be potent and specific inhibitors of TNAP and calf-IALP, respectively, compound 106a showed dual inhibition. The inhibition of b-TNAP and IALP in (µM) 2.41 ± 0.05 and 0.67 ± 0.07, 0.37 ± 0.01 and 40.37%, and 45.31% and 2.90 ± 0.11 µM, respectively, is shown by the 106a, 106b, and 106c. SAR has been conducted using MDS for the active components of the series (Fig. 21) [132].

Oxathiol-2-ylidene-, chromeno indoline–, and sulfonamide-derived inhibitors
Chromeno indoline derivative–based inhibitors
The development of new tetrahydro-6H-spiro[4,3–b] and 10,10-dimethyl-9,10,11,11a-tetrahydrochromoline-7,3′-indoline by using p-toluenesulfonic acid as a catalyst to produce triones (107 and 108). ALP inhibition and prostate cancer treatment properties were identified in these pharmaceutically significant substances (107, 108). When 107 and 108 interacted with human ALP, the selective activity relationship between ALP and prostate cancer was revealed (Fig. 21) [133].
Sulfonamide derivative–based inhibitors
The cyclic sulfonamides (109) were synthesized and assessed for their ability to suppress ALP. The majority of these compounds also showed selective b-TNAP inhibition over bovine IALP inhibition. All the compounds were found to have good inhibitory activity against bTNAP (IC50 = 0.11–6.63 µM). The para-nitro-derivative 109a (IC50 = 0.11 ± 0.005 µM) was the most potent b-TNAP inhibitor. The derivative 109b had the highest level of activity among bovine IALP inhibitors (IC50 = 0.38 ± 0.021 µM). Since the PDB does not provide the crystal structures of bovine ALPs, homology models were created, verified, and then utilized for molecular docking experiments to determine the structural components required for ALP inhibition [134]. The synthesis of hybrids between chalcone and sulfonamide (110) was reported and their assessment as ALP isozyme inhibitors. Maximum inhibition of human and r-CD73 was demonstrated by compounds 110a and 110b, with IC50 ± SEM = 0.26 ± 0.01 and 0.33 ± 0.004 µM, respectively. Furthermore, these compounds were shown to be the calf-IALP-specific inhibitors on ALPs. Maximum inhibition of calf-IALP was shown by derivative 110c, with an IC50 ± SEM = 0.12 ± 0.02 µM [135]. M. Al-Rashida et al. synthesized chromone containing sulfonamides 111 and assessed the inhibitory concentration against ALP. Every drug exhibited exceptional and targeted inhibition of IALP over TNAP, with a Ki value of up to 0.01 ± 0.001 µM. To investigate the specific inhibition exhibited by these drugs, molecular docking investigations were conducted. The most potent IALP inhibitors were discovered to be compounds 112 (Ki, 0.021 ± 0.007 µM) and 111a (Ki, 0.01 ± 0.001 µM) [136]. In order to test the inhibitory efficacy against b-TNAP and IALP, a series of carboxamide-linked aromatic benzenesulfonamides 113 and their sulfonamide-linked bioisosteres 114 were produced. Several of these substances were revealed to be exceptionally strong and specific ALP inhibitors. It was discovered that compound 114a selectively inhibited b-IALP, whereas compound 113a selectively inhibited b-TNAP. Comprehensive kinetic investigations showed a competitive mechanism of inhibition against tissue TNAP and a non-competitive mode of inhibition against IALP for the most active ALP inhibitor 114a. Important binding site interactions were rationalized using molecular docking experiments [137]. The creation of ALP inhibitors is based on sulfonamide 115. They discovered that derivative 115a is a strong TNAP inhibitor that has good plasma levels following subcutaneous dosage, attractive ADME profiles, high selectivity over other ALP, and acceptable water solubility. Furthermore, lead validation investigations also identified derivative 115b, a similarly effective inhibitor (119 nM) from this series. In target validation studies, these compounds may prove useful in assessing the therapeutic potential of TNAP inhibitors for vascular calcification (Fig. 21) [138].
Acetamide/benzamide derivative–based inhibitors
The compounds of acetamides and acetates 116 were produced and assessed as inhibitors of ALP. Based on ALP inhibitory kinetics, compound 116a had the maximum efficacy with an IC50 value of 0.420 ± 0.012 µM, whereas the reference compound (KH2PO4) had an IC50 value of 2.80 µM, suggesting a non-competitive mechanism of interaction with the enzyme. Molecular docking experiments against the ALP enzyme (1EW2) showed that 116a had a good binding affinity with a binding energy value of − 7.90 kcal/mol, when contrasted to other derivatives. The brine shrimp viability testing findings indicated that derivative 116a was safe to employ at the level needed for the enzyme assay. The lead compound 116a had an LD50 of 106.71 µM, while the standard potassium dichromate had an LD50 of 0.891 µM. In an experimental context, spectrophotometric and electrochemical methods were employed to examine the DNA binding contacts of the generated compound 116a. Compound 116a has a strong binding to DNA grooves, as demonstrated by the binding constant values of 7.83 × 103 and 7.95 × 103 M−1, respectively, obtained from UV-Vis spectroscopy and cyclic voltammetry. Since the outcomes of the dry and wet laboratories were in agreement with one another, it was determined that produced compounds, in particular compound 116a, may serve as lead compounds to build the most potent inhibitors of human ALP [139]. The compound benzamide derivative 117 was produced and assessed for their ability to inhibit ALP. With an IC50 value of 0.420 μM, compound 117a had the most powerful action, while the standard (KH2PO4) had an IC50 value of 2.80 μM. To test the synthetic compound 117 binding affinities against the target protein, molecular docking experiments were performed against the ALP enzyme. Three compounds, 117a, 117b, and 117c, had maximal binding interactions with binding energy values of − 8 kcal/mol, according to the docking studies. With a binding distance of 2.13 Å, the molecule 117a demonstrated the interactions between the nitrogen of the oxadiazole ring and the amino acid His265 [140]. The bi-heterocyclic benzamides (118) were tested for their inhibitory actions against ALP, and each of these compounds was demonstrated to be extremely powerful in parallel to the standard. With a potency of 0.0427 ± 0.0167 μM, molecule 118a, which has an aryl component containing a 4-ethoxy group, was found to be the most powerful derivative in the series. Using a Lineweaver–Burk plot, it was demonstrated that 118a had a 1.15 μM Ki value and inhibited ALP uncompetitively. Good interaction behavior inside the target protein's active region was also demonstrated by 118a binding profile (Fig. 22) [141].

Acetamide/benzamide-, thiourea-, and aminoalkanol derivative–based inhibitors
Thiourea derivative–based inhibitors
The investigation looked at the 1,3,4-oxadiazole (122) derivative of salicylic acid, 1-aroyl-3-aryl thiourea (121), bis(thiourea) derivatives of pimelic acid (119), and 3,5-dimethyl pyrazole (120). Compound 119, one of the bis(thiourea) derivatives, exhibited a higher inhibitory activity for h-TNAP, as demonstrated by its IC50 value of 4.63 ± 0.31 µM, which is about four times higher than that of levamisole, the positive control (IC50 value, 19.2 ± 0.01). Compounds 119a, 119b, and 119c had a greater degree of selective inhibition for h-TNAP, as evidenced by their respective IC50 values of 15.4 ± 0.75 µM, 5.28 ± 0.51 µM, and 15.9 ± 0.31 µM. Compound 121 had the highest activity and selectivity for h-IALP, with an IC50 value of 1.50 ± 0.24 µM, when salicylic acid derivatives were compared to the positive control (L-phenylalanine: 80.1 ± 0.01 µM). Compounds 122 and 123 exhibited h-TNAP inhibition with an IC50 value of 4.89 ± 0.84, a value comparable to the inhibitory potential of levamisole, a common inhibitor [142]. A group of acyl/aryl thioureas generated from sulfadiazine 123 was assessed as ALP inhibitors. In the series, compound 123a showed more promise, with an IC50 of 0.251 ± 0.012 µM (compared to the conventional KH2PO4 of 4.317 ± 0.201 µM). The most powerful derivative, 123a, inhibited CIAP via a mixed type of route, according to Lineweaver-Burk plots. Pharmacological analyses revealed that synthetic substances 123 adhere to Lipinski’s law of five. According to the analysis of ADMET parameters, these molecules exhibit substantial lead-like characteristics with the least amount of toxicity and can be used as models for the creation of new drugs (Fig. 22) [143].
Aminoalkanol derivative–based inhibitors
B. Grodner et al. explained the inhibitory effect of aminoalkanol derivatives 124 on the enzymatic TNAP with the use of capillary zone electrophoresis to evaluate the inhibitory effect. Using this method, the quantities of the substrate and product in the reaction mixture with derivatives (124a) or (124b) present may be measured in order to study the enzymatic kinetics. Investigations were conducted into the effects of substituting the dimethylamine group for the propylamine group on the TNAP inhibitory activity of derivatives (124a) and (124b). It was discovered that the substances (124a and 124b) under examination were TNAP inhibitors. Comprehensive kinetic analyses revealed that compound (124a) exhibited a competitive way of inhibition against TNAP, while compound (124b) displayed a non-competitive form of inhibition (Fig. 22) [144].
Quinoline derivative–based inhibitors
The synthesized 4-quinolone derivatives 125 were assessed for their ability to inhibit ALP isozymes. Most of the drugs show moderate selectivity and good inhibitory efficacy. The IC50 results on IALP ranged from 1.06 ± 0.32 to 192.10 ± 3.78 µM, whereas the IC50 values on TNAP were 1.34 ± 0.11 to 44.80 ± 2.34 µM. When compared to TNAP, the most active derivative shows a strong inhibition on IALP with a selectivity that is about 14 times greater. Additionally, to illustrate the binding interactions of the strongest inhibitors inside the active sites of the corresponding enzymes, MDS were carried out [145]. The derivatives of quinolone 125 were demonstrated to be potential TNAP and IALP inhibitors when they were produced by cyclizing α,β-ynones with primary amines. A notable activity against b-TNAP was shown by all aminoquinolones in the range of IC50 ± SEM = 1.14 ± 0.65 to 78.1 ± 1.56 µM. While some of these derivatives were also shown to be potent against calf-IALP, the majority of these derivatives were discovered to be selective inhibitors of b-TNAP. A thorough structural analysis revealed that an aromatic ring at quinolone position 2 is present in all compounds exhibiting strong anti-TNAP action. The bioactivity was diminished when an alkyl group was present. The activity against calf-IALP ranged from IC50 ± SEM = 176.4 ± 2.34 µM to 0.443 ± 0.002. According to the SAR, the inhibitory activity against C-IALP is significantly increased when a hydrophobic or bulky group is present at the nitrogen atom. The most powerful derivative, compound 126a, has IC50 ± SEM = 0.443 ± 0.002, indicating its potency. A chromene substructure seen in other compounds in the series, such as 126b, has been reported to be more potent against c-IALP than against b-TNAP. The inhibitory values measured IC50 ± SEM were 0.797 ± 0.01 against calf-IALP. Conversely, these compounds demonstrated IC50 ± SEM = 5.84 ± 0.99 to 40.9 ± 1.23 µM against b-TNAP [146]. The derivatives of quinoline-4-carboxylic acid 127 were created and assessed for their capability to stop ALP. Most of the substances that were evaluated demonstrated notable inhibition of h-TNAP, tissue-specific h-IALP, and h-PLAP. Of them, 127b stood out as a promising contender against h-IAP and hPLAP, with IC50 values of 34 ± 10 and 82 ± 10 nM, respectively, while 127a was shown to be a strong inhibitor of h-TNAP with an IC50 value of 22 ± 1 nM. With an IC50 value of 150 ± 70 nM, 127c was shown to be a very effective inhibitor of human germ cell ALP. Using homology models based on the h-PLAP structure, MDS were used to deduce the potential binding locations of the most powerful inhibitors (Fig. 23) [147].

Some quinoline, sulfonate, and pyrimidone/pyrimidinone inhibitors
Sulfonate derivative–based inhibitors
The inhibitory action of a variety of sulfonates 128 generated from coumarins against h-TNAP and h-IALP was studied. ALP was effectively inhibited by most of the substances. The most active h-IALP inhibitor was found to be compound 128a, with an IC50 value of 1.11 ± 0.15 µM, whereas the most active h-TNAP inhibitor was found to be compound 128b, with an IC50 value of 0.58 ± 0.17 µM. In order to determine the structural components required for ALP inhibition and to justify the most likely binding site interaction between the inhibitors and the ALP enzymes, SAR and MDS analyses were performed. The most significant structural component—though not the only one—that gives compounds in this series their exceptional ALP inhibitory actions has been hypothesized to be the direct interaction of sulfonate oxygen with the Zn+2 ion [148]. Tricyclic coumarin sulphonate 129 was produced and tested for ALP inhibition against h-TNAP and h-IALP. The derivative 129a with IC50 = 0.38 ± 0.01 µM was discovered to be the most potent inhibitor of h-TNAP. 129b, a different derivative, was discovered to be the most potent h-IALP inhibitor (IC50 = 0.45 ± 0.02 µM). It was also discovered that a few of the compounds are very effective ALP inhibitors. To determine which functional groups are in charge of effectively inhibiting ALP isozymes, SAR research was conducted. Docking studies were utilized to rationalize the most plausible binding site interactions between the discovered inhibitors and the targeted enzymes, hence supporting the study (Fig. 23) [149].
Pyrimidone/pyrimidinone derivative–based inhibitors
The derivatives of fluorinated pyrimidone 130 were synthesized and were discovered to be strong, but non-selective, inhibitors of both ALP isozymes. Compared to h-IALP, which exhibits an IC50 ± SEM = 0.89 ± 0.07 µM, compound 130a exhibits substantial h-TNAP inhibition (IC50 ± SEM = 0.29 ± 0.03 µM). It was shown that compounds with 2-methoxy or 2-ethoxyphenyl groups selectively inhibited h-TNAP, whereas compounds with 4-ethyl or 4-trifloromethoxyphenyl groups selectively inhibited h-IALP. The inhibitory effects of compounds 130b (h-TNAP; 0.21 ± 0.02 µM, h-IALP; 0.43 ± 0.07 µM) and 130c (h-TNAP; 0.28 ± 0.02 µM, h-IALP; 0.48 ± 0.02 µM) were almost equal for both ALPs. The effective inhibitory potential of compounds 130d and 130e was found to be more improved than that of compound 130f (IC50 ± SEM = 1.06 ± 0.05 µM). MDS detected the binding mechanisms and potential interactions of the most active inhibitor inside the enzyme’s active site [150]. The calf ALP test was used to assess the inhibitory impact of dihydropyrimidinone derivatives 131 on ALP. To determine the binding mechanism of active drugs, in silico molecular docking and MDS were used. Compound 131a significantly inhibited the enzyme in the calf ALP inhibitory test, with an IC50 of 1.27 μM at 0.1 µM concentration, compared to standard KH2PO4, which had an IC50 of 2.80 μM. At the same concentration, compounds 131b, 131c, and 131d likewise demonstrated extremely good inhibition, with IC50 values of 2.502, 2.943, and 2.132 μM, respectively. Compounds 131b, 131c, and 131d had effective radical scavenging activity at 100 μg/mL, with IC50 values of 0.48, 0.61, and 0.75 μg/mL, respectively, according to the antioxidant test. Effective binding of active drugs at the target enzyme’s active binding site was demonstrated by the MDS investigations. Good predictivity and statistical validation were found in the final QSAR equation, with R2 = 0.958 and Q2 = 0.903 for the developed model, respectively. With consistent binding modes, compound 131a demonstrated the strongest inhibitory efficacy and might serve as a potential lead for the discovery of ALP inhibitors (Fig. 23) [151].
Chalcone/aurone- and benzothiazine derivative–based inhibitors
The ability of the chalcone and 1,2-benzothiazine derivatives 132 to inhibit the ALP isoforms h-TNAP and h-IALP was studied. All the para-substituted compounds exhibited modest selectivity, but they were active against both isoforms in the low micromolar dose range. Compound 132a stands up as the most effective inhibitor of h-TNAP, with a selectivity index of 0.1 and a potency of 0.25 µM. High selectivity for h-IALP was reported for the meta-substituted compounds 132b, 132c, and 132d, with 132b being the most effective at 1.04 µM. In the active site of the two ALP isoforms for h-IALP and h-TNAP, respectively, MDS identified distinct interaction mechanisms for compounds 132b and 132a [152]. A derivative of aurone 134 and chalcone 133 was assessed as strong ALP inhibitors. Compounds 134a (IC50 = 2.163 ± 0.048 µM), 134b (IC50 = 2.146 ± 0.056 µM), 134c (IC50 = 2.132 ± 0.034 µM), 134d (IC50 = 1.154 ± 0.043 µM), 134e (IC50 = 1.055 ± 0.029 µM), and 134f (IC50 = 2.326 ± 0.059 µM) displayed outstanding inhibitory activity against ALP, outperforming/being even more active than KH2PO4 (standard) (IC50 = 2.80 ± 0.065 µM). Interestingly, compound 134e may be used as a model structure to create ALP inhibitors with higher potencies. Surprisingly, compound 134e might be used as a model structure to create ALP inhibitors with higher potencies. Compound 134e was identified as a putative ALP inhibitor after MDS testing was done to assess the compounds’ dynamic behavior, protein–ligand complex stability, and binding affinity. The evaluation of the ADMET parameters revealed that these substances have many properties similar to lead, are low in toxicity, and may be used as models for the creation of new drugs (Fig. 24) [153].

Chalcone/aurone and benzothiazine-, okadaic acid–, catechol-, coumarin–triazolothiadiazine-, and furan derivative–based inhibitors
Okadaic acid derivative–based inhibitors
V. Meštrovic et al. studied the SAR of okadaic acid with ALP and consider that compound have ability to inhibit the protein phosphatase. Compound 135 functions as a non-competitive inhibitor of ALP, according to kinetic study of ALP from Escherichia coli, human placental, and calf-IALP. Compared to the eukaryotic proteins (human placental ALP, Ki 2.05 µM; calf-IALP, Ki 3.15 µM), the bacterial enzyme has a greater affinity for compound 135 (Ki 360 nM). Through control of the phosphorylation/dephosphorylation balance of proteins containing phosphoseryl or phosphothreonyl residues, ALP may have a role in the phosphorylation state, as shown by the inhibition by compound 135 (Fig. 24) [154].
Catechol derivative–based inhibitors
ALP was inhibited by a series of 3,4-dihydroxy-substituted catechols 136. They discovered that PLAP’s best inhibitor is 136a. PLAP inhibitors have a higher degree of inhibitory selectivity against TNAP and IALP. The compound 136a showed better inhibiting selectivity for PLAP than that of TNAP and IALP because of molecular alteration. Compared to TNAP and IALP, compound 136b, which has a 2-ethylimidazole substituent, was nearly 27 times more selective as a PLAP inhibitor while maintaining a respectable level of PLAP inhibitory efficacy (IC50 = 4.2 µM). When it came to inhibiting PLAP over TNAP and IALP, compound 136c was more than 50- and 25-fold selective, respectively. Ultimately, compared to IALP and TNAP, compound 136d was a 10- and 40-fold more selective inhibitor of PLAP. 136c, 135b, and 136d, the three compounds, were shown to be more effective PLAP inhibitors than previously documented isozyme-selective ALP inhibitors (Fig. 24) [155].
Coumarin–triazolothiadiazine derivative–based inhibitors
After coumarin–triazolothiadiazine hybrid compounds 137 were assessed against ALP, compound 137a—which incorporates bis-coumarinyl motifs at the heteroaromatic core’s 3- and 6-positions—proved to be a strong inhibitor, with an IC50 value of 1.15 ± 1.0 µM. Additionally, the created compounds were evaluated against Leishmania major, with 137b demonstrating the highest potency with an IC50 value of 0.89 ± 0.08 µM. Compound 137c exhibited superior cytotoxic potential against H-157 cells, with an IC50 value of 1.01 ± 0.12 µM. This represents an enhanced inhibition when matched to the standards (cisplatin and vincristine) utilized in the experiment. The synthesized library of coumarin–triazolothiadiazine hybrids was subjected to MDS testing against ALP. Nearly all of the substances demonstrated strong interactions with the essential residues of the receptor’s active site (Fig. 24) [156].
Furan derivative–based inhibitors
The compound 3-(3-arylprop-2-ynyl)dihydrofuran-2(3H)-one (138) was shown to be an ALP inhibitor. Significant and specific TNAP inhibitors were discovered in the majority of compounds. While compound 138b had 104 times more inhibitory capability on c-IALP when compared to reference L-phenylalanine, compound 138a demonstrated 14 times more inhibition against b-TNAP when compared to levamisole. In order to address vascular calcification, potent and targeted b-TNAP inhibitors may be beneficial. Potent inhibitors’ binding mechanisms inside the active pocket of each enzyme were further clarified by the docking studies conducted for the tested drugs (Fig. 24) [157].
Acridine derivative–based inhibitors
The synthetic acridine derivative 139 was studied as an ALP inhibitor. Because of their distinctive conjugated planar heterocyclic structure, which strongly intercalates with ALP, acridine analogs have a strong inhibitory potential. IC50 = 0.0102 ± 0.0005 µM for analog 139a illustrated the greatest capability in the sequence associated to conventional KH2PO4 = 4.317 ± 0.201 μM (Fig. 25) [158].

Some acridine-, trizole-, thiazole-, and thiadiazole derivative–based inhibitors
Trizole-, thiazole-, and thiadiazole derivative–based inhibitors
A series of 2,5-disubstituted-1,3,4-thiadiazole derivatives (141) and 4,5-disubstituted-2,4-dihydro-3H-1,2,4-triazole-3-thione derivatives (140) were synthesized and assessed as inhibitors of ALP as well as acetylcholinesterase. When compared to the standard drug, the majority of the assessed derivatives demonstrated capable actions. Of these, compounds (140a) and (140b) demonstrated excellent acetylcholinesterase inhibitory activity with IC50 = 0.241 ± 0.012 and 0.260 ± 0.013 µM, respectively. The most potent ALP inhibitors were compounds (140c) with IC50 = 0.044 ± 0.001 µM and 141a with IC50 = 0.15 ± 0.02 µM and Ki 0.11 ± 0.02 µM [159]. The inhibitory power of ALP on synthetic thiazole-linked thioureas with aliphatic and aromatic side chains (142) was evaluated. They claim that while the synthesized compounds are strong ALP inhibitors, the best compounds had the lowest IC50 values—0.057 and 0.019 µM, respectively—for 142a and 142b. The compounds 142a and 142b had the greatest docking energies of − 32.18 and − 30.09 kJ/mol, respectively, out of all the compounds. According to the findings, these compounds may be employed in the future to create more powerful ALP inhibitors that will be used to treat a variety of cancers, including breast cancer [160]. The ability of azomethine-clubbed thiazoles (143) to inhibit h-TNAP and h-IALP was evaluated. With IC50 values of 0.15 ± 0.01 and 0.50 ± 0.01 µM, respectively, compounds 143a and 143b were determined to be the most powerful for h-TNAP, whereas compounds 143c and 143b showed the highest potency for h-IALP, with IC50 values of 2.59 ± 0.04 and 2.56 ± 0.02 µM, respectively. MDS were also used to determine the kind of binding contact that may exist between an inhibitor and an enzyme’s active site. To determine the mechanism of enzyme inhibition, kinetic investigations of enzyme inhibition were conducted [161]. The ALP inhibitory potential of substituted hydrazine derivatives 144 was examined. With an IC50 value of 1.09 ± 0.18 µM, compound 144a was shown to be the most effective h-TNAP inhibitor among this group of compounds. For h-IALP, compound 144b demonstrated efficacy and selectivity with an IC50 value of 0.71 ± 0.02 µM. Furthermore, SAR and MDS were used to assess how well they bound to the ALP target location. According to the docking research, the most potent inhibitors had significant contacts inside the h-TNAP and h-IALP binding pockets, which may account for the compound’s inhibitory impact on the enzymes (Fig. 25) [162].
Chromone derivative–based inhibitors
ALP-inhibitors were assessed for 3,3ʹ-carbonyl-bis(chromones) 145, which are dimeric chromones connected by a carbonyl group. With an IC50 value of 2.47 ± 0.003 µM, 145a was concluded to be the efficient inhibitor of b-TNAP among all the compounds that were studied. Compared to the reference chemical levamisole, which had an IC50 value of 19.21 ± 0.001 µM, it exhibited a ninefold more inhibitory potential. With an inhibitory value of IC50 = 0.653 ± 0.003 µM, compound 145b was shown to be the most effective inhibitor against calf-IALP. Compared to the reference inhibitor L-phenylalanine, which had an IC50 value of 80.21 ± 0.001µM, it was more than 120 times more powerful. The thorough structural analysis supported the theory that this compound’s action might be attributed to benzene and carboxamide directly attaching to the chromene core ring. When the activity of this compound was tested with other derivatives having two chromene rings, it became clear that the compound with one 4H-chromen-4-one ring was more active against b-TNAP than the compound with two 4H-chromen-4-one rings (Fig. 26) [163].

Some chromone-, pyridine-, thiazoline-, and propanamide-based inhibitors
Pyridine derivative–based inhibitors
The h-TNAP enzyme was employed in the testing of the pyridine and dihydropyridine derivatives 146. With IC50 values ranging from 0.49 ± 0.025 to 8.8 ± 0.53 µM, most of the compounds showed excellent h-TNAP-specific enzyme inhibition. This contrasts with the conventional inhibitor of the h-TNAP enzyme, levamisole, which has an IC50 value of 22.65 ± 1.60 µM. To look into proapoptotic activity, the most powerful dihydropyridine-based analog, 146a, was chosen. Using flow cytometry, microscopy, and staining agents, the apoptosis-inducing activity of compound 146a was investigated. To determine the primary structural elements preventing the h-TNAP enzyme from enzymatically activating, comprehensive SAR and MDS analyses were conducted (Fig. 26) [164].
Thiazoline derivative–based inhibitors
The potential for ALP inhibition was assessed for iminothiazoline derivative 147, which is based on quinolinyl. In comparison to other synthesized derivatives and reference compound KH2PO4 (IC50 = 5.245 ± 0.477 µM), compound 147a demonstrated the highest ALP inhibitory action (IC50 = 0.337 ± 0.015 µM). The derivatives (147a), according to kinetic studies, had a Ki value of 0.47 mM and was a non-competitive inhibitor of alkaline phosphatase. Compound 147a is an excellent inhibitor of the pointed protein ALP, as demonstrated by molecular docking. Comprehensive MDS were conducted to assess docking data validity further, confirming 147a’s inhibitory capacity. Iminothiazolines’ quinolinyl and aryl or alkyl moiety significantly affects their ability to inhibit ALP. The derivative 147a might be used to create a medication with greater potency for the suppression of ALP [165]. A. Ahmed et al. synthesized and studied SAR of 1,3-thiazolines derivatives derivatives 148 against ALP. DFT simulations were performed to evaluate the electronic characteristics, and the molecular docking tool was used to examine the binding interactions of the synthesized derivatives. The assessment of the bioactivity of produced compounds against ALP yielded encouraging findings. It was within acceptable bounds for the drug likeliness score, which is a signal for any chemical entity masquerading as a drug. According to the results, the majority of the derivatives were strong ALP inhibitors, which might assist as lead compound for the creation of derivatives with the appropriate pharmacological profiles for the treatment of particular disorders linked to aberrant ALP levels (Fig. 26) [166].
Propanamide derivative–based inhibitors
ALP inhibition was examined using a series of propanamide derivatives, 149. As can be seen from their decreased IC50 (μM) values compared to the reference, KH2PO4, which had an IC50 value of 5.242 ± 0.473 μM, all compounds showed extremely good activity. Propenamides 149 competitively inhibited this enzyme by creating an enzyme-inhibitor complex, according to the kinetic mechanism examined by Lineweaver-Burk plots. Additionally, these substances were examined for their cytotoxic properties using hemolytic activity, and it was found that almost all of these propanamides showed minimal cytotoxicity. The mechanism of action explains why compound 149a is the most effective in inhibiting ALP, with an IC50 value of 0.531 ± 0.003 μM (Fig. 26) [167].
Dual inhibitors
Nucleotide-based inhibitors
According to Jeffrey et al., several clinical trials are being conducted to assess the effectiveness of anti-CD73/CD39 mAbs for the treatment of cancer, either on their own or in conjunction with other proven medications. A number of small-molecule CD73 inhibitors with enhanced potency, selectivity, and drug-like qualities have recently been found as a result of growing interest in the adenosine signaling system. AB680 (150), the first small-molecule CD73 inhibitor to reach clinical trials, is among these inhibitors. Compound 150 is a highly selective and powerful (Ki = 5 pM) inhibitor of CD73 that is now being tested in combination regimens to treat metastatic castration-resistant prostate cancer and advanced pancreatic cancer. It was recently determined that compound 150 is safe and well-tolerated in people [168]. Sulfopolysaccharides from brown and red sea algae have been shown to function as strong dual inhibitors of ENPP1 and NTPDase1, CD39, the primary ATP-hydrolyzing ectoenzymes. These inhibitors exhibit nano- to picomolar efficacy and a non-competitive manner of inhibition. They demonstrated how, in a concentration-dependent way, one of the sulfopolysaccharides examined as a sample example decreased the synthesis of adenosine at the surface of the human glioblastoma cell line U87. These natural substances have the potential to be innovative treatments for cancer immunotherapy since they are the most powerful inhibitors of extracellular ATP hydrolysis yet identified [169]. Schäkel and colleagues developed derivatives and analogs of ARL67156 (151), a nucleotide analog that exhibits a competitive method of inhibition, the standard CD39 inhibitor. Replacements in the N6 and C8 positions of the adenine core as well as changes to the triphosph(on)ate chain were examined in SAR analysis at the human enzyme. Of the current series, compounds 151 and its variants 151a and 151b, which had Ki values of about 1 µM, were the most effective inhibitors of CD39/CD73. All three nucleotide analogs functioned as dual-target inhibitors by blocking CD73, as evidenced by selectivity experiments. Realistic binding modalities to both targets were given by docking experiments (Fig. 27) [170].

Nucleotide-based dual inhibitors
Non-nucleotide-based inhibitors
Sulfonamide- and sulfonylhydrazone derivative–based CD73 and ALP inhibitors
The most effective inhibitor for h-TNAP and h-IALP was found to be the chromen-2-one scaffold-based sulfonylhydrazones 152, with IC50 values of 1.02 ± 0.13 and 0.32 ± 0.03 µM, respectively, when compared to levamisole (IC50 = 25.2 ± 1.90 µM for h-TNAP) and L-phenylalanine (IC50 = 100 ± 3.00 µM for h-IALP) as standards. Moreover, compound 153 based on chromen-2-one exhibited remarkable activity against CD73 with an IC50 value of 0.29 ± 0.004 µM, compared to standard sulfamic acid (IC50 = 42.1 ± 7.8 µM). Out of the series of sulfonylhydrazones based on phenyl rings, compound 154 was shown to be the most effective against h-TNAP and h-IALP, with IC50 values of 0.85 ± 0.08 and 0.52 ± 0.03 µM. Furthermore, in silico studies were carried out to demonstrate their possible affinity with the target enzymes. In order to develop innovative therapeutic medications, strong compounds 152, 153, and 154 against different ectonucleotidases (CD73, h-TNAP, and h-IALP) may be utilized as a model [171]. Several chromone sulfonamides (155) and sulfonylhydrazones (156, 157) were assessed for their ability to suppress human and rat CD73 as well as human ALP (h-TNAP and h-IALP). Compound 157a was discovered to be a very potent and selective h-TNAP inhibitor (h-TNAP IC50 = 1.41 ± 0.10 µM; h-IALP = 43.1%), whereas compound 155a had the maximum activity as an h-IALP inhibitor (h-IALP IC50 = 0.51 ± 0.20 µM; h-TNAP = 36.5%). 156a has the highest level of activity as a CD73 inhibitor (IC50 = 0.18 ± 0.02 µM). MDS suggest that among all the non-bonded contacts in the enzyme’s active site, the one between the sulfonamide group and the Zn atom may have been the most significant (Fig. 28) [172].

Sulfonamide-, sulfonylhydrazone-, thienotetrahydropyridine-, chalcone sulfonamide–, and pyridine derivative–based dual inhibitors
Thienotetrahydropyridine derivative–based CD39 and CD73 inhibitors
The compound 2‒substituted thienotetrahydropyridine derivatives 158, which have structural similarities to ticlopidine, were examined as CD39 inhibitors. They can be anticipated to have no P2Y12 receptor-antagonistic action in vivo because of their substituent on the 2‒position, which prevents them from being metabolically changed into reactive thiols. Many thienotetrahydropyridine compounds (158) inhibited CD39 in a concentration-dependent manner. As an allosteric inhibitor, ticlopidine and its most powerful derivative, 158a, both demonstrated comparable CD39-inhibitory efficacy. 158a was identified as a new dual inhibitor of CD39 and CD73, whereas ticlopidine inhibited many NTPDase isoenzymes (Fig. 28) [173].
Chalcone sulfonamide–based CD73 and ALP inhibitors
The inhibitory potential of chalcone-sulfonamide hybrids 159 and their derivatives was assessed in relation to two ectonucleotidase family members, CD73 and ALP. It was discovered that only six compounds could block the rat and human CD73 enzymes. Most inhibition of h-CD73 and r-CD73 was demonstrated by compounds 159a and 159b, with IC50 ± SEM = 0.26 ± 0.01 and 0.33 ± 0.004 µM, respectively. Furthermore, these compounds were shown to be the calf IALP’s specific inhibitors on ALP. Derivative 159c demonstrated the highest inhibition of calf-IALP, with an IC50 ± SEM = 0.12 ± 0.02 µM. In conclusion, these chalcone-sulfonamide hybrids were more selective for the calf-IALP enzyme yet demonstrated dual inhibition of both isozyme families (Fig. 28) [174].
Pyridine derivative–based ALP and CD73 inhibitors
Hassan et al. reported the synthesis of 4-aminopyridine derivative 160, which was then assessed using detailed SAR as an inhibitor of ALP and CD37. Compound 160a demonstrated significant inhibition (IC50 ± SEM = 0.25 ± 0.05 µM), which was found to be 168 times more potent than the previously reported inhibitor suramin (IC50 ± SEM = 42.1 ± 7.8 µM). The selectivity of this chemical towards hTNAP was six times higher than that of CD73. These compounds also showed anti-cancer potential which were studied using cell viability assay, flow cytometric analysis and nuclear staining. To learn more about the binding interactions of strong substances within the corresponding enzyme compartments and herring-sperm DNA, MDS tests were also performed (Fig. 28) [175].
Pyrazolyl pyrimidinetrione derivative–based ALP and ENPP inhibitors
The ability of the pyrazolyl pyrimidinetriones and thioxopyrimidinediones 161 to inhibit human ALP (h-TNAP and h-IALP) and ectonucleotidase (h-ENPP1 and h-ENPP3) enzymes was assessed. Depending on the functionalized hybrid structure, most of the evaluated compounds exhibited varying degrees of inhibition, making them extremely powerful. The comprehensive surface area ratio analysis of 161 derivatives revealed that compound 161b selectively inhibited the h-IALP isozyme with an IC50 value of 0.86 ± 0.04 µM, whereas compound 161a, which has an unsubstituted phenyl ring, resulted to a powerful and selective inhibition of h-TNAP (IC50 = 0.33 ± 0.02 µM). Similarly, the lead scaffolds against h-ENPP1 and h-ENPP3, respectively, were found to be compounds 161c and 161d. The most effective inhibitors’ likely binding mechanisms were determined using molecular docking analysis (Fig. 29) [176].

Pyrazolyl pyrimidinetrione–, coumarin-, isonicotinohydrazone-, deazapurine-, and chromanones derivative–based dual inhibitors
Coumarin derivative–based CD73 and ALP inhibitors
The ability of each molecule in the series of 2H-chromen-2-one derivatives 162 to stop human recombinant ectonucleotidases, such as h-TNAP and h-IALP, as well as human and rat CD73, was assessed. Compounds 162a (IC50 = 0.25 ± 0.07 µM) and 162b (IC50 = 0.28 ± 0.05 µM) were the most potent h-CD73 inhibitors. Compounds 162c and 162d demonstrated the strongest inhibition of h-TNAP (IC50 ± 0.21 0.04 µM and 0.22 ± 0.03 µM, respectively). This is about 91 times more inhibiting than the conventional inhibitor levamisole. Compound 162e (IC50 = 0.05 ± 0.001 µM) was shown to be the most efficient h-IALP inhibitor, exhibiting ≈ 11 times greater selectivity for h-IALP than h-TNAP. Strong inhibitors and ectonucleotidases were shown to have the most probable binding site interactions using molecular docking, dynamics studies, and homology modeling (Fig. 29) [177].
Isonicotinohydrazone derivative–based CD73 and ALP inhibitors
A variety of compounds of isonicotinohydrazide (163–164) was synthesized and evaluated against rat and human recombinant CD73 and ALP isozymes, including tissue-specific calf IALP and b-TNAP. Vascular calcifications, hypophosphatasia, solid tumors, and malignancies of the colon, lung, breast, pancreas, and ovary are all associated with these enzymes. Every tested substance showed activity against both enzymes. Derivative 163a showed substantial inhibitory effect against r-CD73, whereas derivative 164 was the most powerful inhibitor of CD73. Furthermore, the derivative 163a was shown to be the strongest inhibitor of b-TNAP, whereas the derivative 163b was the most potent inhibitor against calf-IALP. Additionally, potential binding mechanisms of strong drugs (such as b-TNAP and c-IALP) against rat and human CD73 and ALP were computationally ascertained (Fig. 29) [178].
Deazapurine derivative–based ALP and ENPP inhibitors
In order to determine the compounds’ inhibitory efficacy against human recombinant ALP and ENPP enzymes, a synthesis of fluorinated and non-fluorinated 1H-pyrazolo[3,4-b]pyridin-3-ones 165 was conducted. A strong and specific inhibition of both target enzymes was found in the findings of an in vitro biological experiment. The most specific inhibition of h-TNAP was demonstrated by compound 165a, whereas h-IALP isozyme was exclusively inhibited by compound 165b. It is noteworthy that compounds 165c and 165d were shown to be effective lead scaffolds against human ENPP1 and ENPP3, respectively. According to the docking data, the molecules engage with the Zn ion and the essential amino acid residues through hydrogen bonds and π–π interactions (Fig. 29) [179].
Chroman-4-one derivative–based ALP and CD73 inhibitors
It was discovered that 2-alkoxy-3-(sulfonylarylaminomethylene)-chroman-4-ones 166 were specific inhibitors of IALP, TNAP, and CD73. Comprehensive analyses of enzyme kinetics demonstrated non-competitive inhibition against human and rat CD73 and competitive inhibition against ALP. Compared to IALP (Ki = 2.18 ± 0.12 µM), the most potent TNAP inhibitor 166a (Ki = 0.078 ± 0.001 µM) has shown 28 times more selectivity for TNAP. At 300 times more selective for IALP than TNAP (Ki = 72.9 ± 1.68 µM), compound 166b was the most potent inhibitor of IALP (Ki = 0.24 ± 0.01 µM). The most effective human ecto-50 nucleotidase inhibitor was compound 166c, which showed inhibition in the low nanomolar range (Ki = 14 nM) (Fig. 29) [180].
Conclusions
Ectonucleotidases are a family of cell surface enzymes that are crucial for the regulation of adenosine and external nucleotide signaling. The extracellular microenvironment’s pro-inflammatory ATP to anti-inflammatory adenosine ratio is regulated by these enzymes, particularly CD39 and CD73. Ectonucleotidase inhibitors, a broad family of compounds, have become effective modulators of purinergic signaling pathways. In this comprehensive review, we examine the varied terrain of ectonucleotidase inhibitors, classify them into different kinds, and analyze their modes of action. We explore the complex network of biological processes that these inhibitors affect, from immunological control and inflammation to the development of cancer and cardiovascular disease. Additionally, we investigated the ectonucleotidase inhibitors’ therapeutic potential, focusing on their uses in the therapy of autoimmune disorders, cancer immunotherapy, and other clinical situations. Optimizing selectivity, pharmacokinetics, and combination medicines are explored along with the potential and challenges related to their development and clinical translation. Ectonucleotidase inhibitors are at the forefront of precision medicine, offering targeted therapies with the potential to completely alter the landscape of treatment for a wide range of diseases. This is due to the interdisciplinary collaboration between researchers in pharmacology, immunology, and oncology that is currently thriving.
Data availability
No datasets were generated or analyzed during the current study.
Abbreviations
- ATP:
-
Adenosine triphosphate
- NTPDase:
-
Ectonucleoside triphosphate di-phosphohydrolases
- UDP:
-
Uridine-5′-diphosphate
- h-NTPDase:
-
Human NTPDase
- r-NTPDase:
-
Rat NTPDase
- ENPP:
-
Ectonucleotide pyrophosphatase/phosphodiesterases
- h-ENPP:
-
Human ENPP
- ADP:
-
Adenosine diphosphate
- ALP:
-
Alkaline phosphatases
- TNAP:
-
Tissue-nonspecific alkaline phosphatase
- QSAR:
-
Quantitative structure activity relationship
- IALP:
-
Intestinal alkaline phosphatase
- MDS:
-
Molecular dynamics simulations
- b-TNAP:
-
Bovine-tissue non-specific ALP
- BIALP:
-
Bovine intestinal alkaline phosphatase
- NAD+ :
-
Nicotinamide-adenine dinucleotide
- PPADS:
-
Pyridoxalphasposphate-6-azophenyl-2ʹ,4ʹ-di-sulphonic acid
- AMP:
-
Adenosine monophosphate
- RB-2:
-
Reactive blue 2
- STING:
-
Stimulator of interferon genes
- UTP:
-
Uridine-5′-triphosphate
- LOD:
-
Limit of detection
- cAMP:
-
Cyclic adenosine monophosphate
- R 2 :
-
The correlation coefficient
- PC-1:
-
Plasma cell membrane protein-1 (ENPP-1)
- SAR:
-
Structure activity relationship
- CYP:
-
Cytochrome P450
- ADME:
-
Absorption, distribution, metabolism, and excretion
- TME:
-
Tumor microenvironment
- CoMFA:
-
Comparative molecular field analysis
- MD:
-
Molecular dynamics
- Q 2 :
-
Cross-validated coefficient
- VS:
-
Virtual screening
- HUVEC:
-
Human umbilical vein endothelial cells
- hERG:
-
Human ether-a-go-go-related gene
- PLAP:
-
Placental alkaline phosphatase
- MOA:
-
Mechanism of action
- CoMSIA:
-
Comparative molecular similarity index analysis
References
Zimmermann H, Zebisch M, Sträter N (2012) Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal 8:437–502
Fredholm BB, IJzerman AP, Jacobson KA, Klotz K-N, Linden J (2001) International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev 53(4):527–52
Fredholm BB, IJzerman AP, Jacobson KA, Linden J, Müller CE (2011) International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—an update. Pharmacol Rev 63(1):1–34
Al-Rashida M, Qazi SU, Batool N, Hameed A, Iqbal J (2017) Ectonucleotidase inhibitors: a potent review (2011–2016). Expert Opin Ther Pat 27(12):1291–1304
Gao Z-G, Jacobson KA (2007) Emerging adenosine receptor agonists. Expert Opin Emerg Drugs 12(3):479–492
Fredholm BB, Abbracchio MP, Burnstock G, Dubyak GR, Harden TK, Jacobson KA et al (1997) Towards a revised nomenclature for P1 and P2 receptors. Trends Pharmacol Sci 18(3):79
Tozaki-Saitoh H, Takeda H, Inoue K (2022) The role of microglial purinergic receptors in pain signaling. Molecules 27(6):1919
Haskó G, Pacher P (2008) A2A receptors in inflammation and injury: lessons learned from transgenic animals. J Leukoc Biol 83(3):447–455
Nishat S, Khan LA, Ansari ZM, Basir SF (2016) Adenosine A3 receptor: a promising therapeutic target in cardiovascular disease. Curr Cardiol Rev 12(1):18–26
Mahmood A, Iqbal J (2022) Purinergic receptors modulators: an emerging pharmacological tool for disease management. Med Res Rev 42(4):1661–1703
Bao X, Xie L (2022) Targeting purinergic pathway to enhance radiotherapy-induced immunogenic cancer cell death. J Exp Clin Cancer Res 41(1):1–18
Jacob F, Novo CP, Bachert C, Van Crombruggen K (2013) Purinergic signaling in inflammatory cells: P2 receptor expression, functional effects, and modulation of inflammatory responses. Purinergic Signal 9:285–306
North RA (2016) P2X receptors. Philos Trans R Soc B: Biol Sci 371(1700):20150427
Baqi Y (2015) Ecto-nucleotidase inhibitors: recent developments in drug discovery. Mini Rev Med Chem 15(1):21–33
Hechler B, Cattaneo M, Gachet C, editors. The P2 receptors in platelet function. Seminars in thrombosis and hemostasis; 2005: Copyright 2005 by Thieme Medical Publishers, Inc., 333 Seventh Avenue, New York.
Burnstock G (2012) Purinergic signalling: its unpopular beginning, its acceptance and its exciting future. BioEssays 34(3):218–225
Lazarowski ER (2012) Vesicular and conductive mechanisms of nucleotide release. Purinergic Signal 8(3):359–373
Burnstock G (2017) Purinergic signalling and neurological diseases: an update. CNS & Neurological Disorders-Drug Targets (Formerly Current Drug Targets-CNS & Neurological Disorders) 16(3):257–65
von Kügelgen I, Hoffmann K (2016) Pharmacology and structure of P2Y receptors. Neuropharmacology 104:50–61
Frelinger AL, Bhatt DL, Lee RD, Mulford DJ, Wu J, Nudurupati S et al (2013) Clopidogrel pharmacokinetics and pharmacodynamics vary widely despite exclusion or control of polymorphisms (CYP2C19, ABCB1, PON1), noncompliance, diet, smoking, co-medications (including proton pump inhibitors), and pre-existent variability in platelet function. J Am Coll Cardiol 61(8):872–879
Cattaneo M (2015) P2Y12 receptors: structure and function. J Thromb Haemost 13:S10–S16
Abbracchio MP, Burnstock G, Verkhratsky A, Zimmermann H (2009) Purinergic signalling in the nervous system: an overview. Trends Neurosci 32(1):19–29
Haas CB, Lovászi M, Braganhol E, Pacher P, Haskó G (2021) Ectonucleotidases in inflammation, immunity, and cancer. J Immunol 206(9):1983–1990
Nitschke Y, Rutsch F (2012) Genetics in arterial calcification: lessons learned from rare diseases. Trends Cardiovasc Med 22(6):145–149
Umezu-Goto M, Kishi Y, Taira A, Hama K, Dohmae N, Takio K et al (2002) Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J Cell Biol 158(2):227–233
Ferrero E, Faini AC, Malavasi F (2019) A phylogenetic view of the leukocyte ectonucleotidases. Immunol Lett 205:51–58
Johnson RC, Leopold JA, Loscalzo J (2006) Vascular calcification: pathobiological mechanisms and clinical implications. Circ Res 99(10):1044–1059
Choi J (2023) Small molecule ectonucleotide pyrophosphatase/phosphodiesterase 1 inhibitors in cancer immunotherapy for harnessing innate immunity. Bull Korean Chem Soc 44(2):88–99
Antonioli L, Blandizzi C, Pacher P, Haskó G (2013) Immunity, inflammation and cancer: a leading role for adenosine. Nat Rev Cancer 13(12):842–857
Eltzschig HK, Sitkovsky MV, Robson SC (2012) Purinergic signaling during inflammation. N Engl J Med 367(24):2322–2333
Allard B, Longhi MS, Robson SC, Stagg J (2017) The ectonucleotidases CD 39 and CD 73: novel checkpoint inhibitor targets. Immunol Rev 276(1):121–144
Wang L, Fan J, Thompson LF, Zhang Y, Shin T, Curiel TJ et al (2011) CD73 has distinct roles in nonhematopoietic and hematopoietic cells to promote tumor growth in mice. J Clin Investig 121(6):2371–2382
Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK et al (2002) Ecto-5′-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Investig 110(7):993–1002
Neves GM, Kagami LP, Battastini AMO, Figueiró F, Eifler-Lima VL (2023) Targeting ecto-5′-nucleotidase: a comprehensive review into small molecule inhibitors and expression modulators. Eur J Med Chem 247:115052
Bajracharya B, Shrestha D, Talvani A, Gonçalves R, Afonso LCC (2022) The ecto-5 nucleotidase/CD73 mediates Leishmania amazonensis survival in macrophages. BioMed Res Int 2022:9928362
Sträter N (2006) Ecto-5’-nucleotidase: Structure function relationships. Purinergic Signal 2:343–350
Pasquini S, Contri C, Borea PA, Vincenzi F, Varani K (2021) Adenosine and inflammation: here, there and everywhere. Int J Mol Sci 22(14):7685
Savio LE, de Andrade MP, Da Silva CG, Coutinho-Silva R (2018) The P2X7 receptor in inflammatory diseases: angel or demon? Front Pharmacol 9:52
Wiley J, Sluyter R, Gu B, Stokes L, Fuller S (2011) The human P2X7 receptor and its role in innate immunity. Tissue Antigens 78(5):321–332
Baghbani E, Noorolyai S, Shanehbandi D, Mokhtarzadeh A, Aghebati-Maleki L, Shahgoli VK et al (2021) Regulation of immune responses through CD39 and CD73 in cancer: Novel checkpoints. Life Sci 282:119826
Millán JL (2006) Alkaline phosphatases: structure, substrate specificity and functional relatedness to other members of a large superfamily of enzymes. Purinergic Signal 2:335–341
Le-Vinh B, Akkuş-Dağdeviren ZB, Le NMN, Nazir I, Bernkop-Schnürch A (2022) Alkaline phosphatase: a reliable endogenous partner for drug delivery and diagnostics. Adv Ther 5(2):2100219
Sharma U, Pal D, Prasad R (2014) Alkaline phosphatase: an overview. Indian J Clin Biochem 29:269–278
Siede WH, Seiffert UB, Merle S, Goll H-G, Oremek G (1989) Alkaline phosphatase isoenzymes in rheumatic diseases. Clin Biochem 22(2):121–124
Haarhaus M, Brandenburg V, Kalantar-Zadeh K, Stenvinkel P, Magnusson P (2017) Alkaline phosphatase: a novel treatment target for cardiovascular disease in CKD. Nat Rev Nephrol 13(7):429–442
Haarhaus M, Cianciolo G, Barbuto S, La Manna G, Gasperoni L, Tripepi G et al (2022) Alkaline phosphatase: an old friend as treatment target for cardiovascular and mineral bone disorders in chronic kidney disease. Nutrients 14(10):2124
Zaher DM, El-Gamal MI, Omar HA, Aljareh SN, Al-Shamma SA, Ali AJ et al (2020) Recent advances with alkaline phosphatase isoenzymes and their inhibitors. Arch Pharm 353(5):e2000011
Al-Rashida M, Iqbal J (2015) Inhibition of alkaline phosphatase: an emerging new drug target. Mini Rev Med Chem 15(1):41–51
Eliahu S, Lecka J, Reiser G, Haas M, Bigonnesse F, Lévesque SA et al (2010) Diadenosine 5′, 5′′-(boranated) polyphosphonate analogues as selective nucleotide pyrophosphatase/phosphodiesterase inhibitors. J Med Chem 53(24):8485–8497
Zelikman V, Pelletier J, Simhaev L, Sela A, Gendron F-P, Arguin G et al (2018) Highly selective and potent ectonucleotide pyrophosphatase-1 (NPP1) inhibitors based on uridine 5′-Pα, α-dithiophosphate analogues. J Med Chem 61(9):3939–3951
Nadel Y, Lecka J, Gilad Y, Ben-David G, Förster D, Reiser G et al (2014) Highly potent and selective ectonucleotide pyrophosphatase/phosphodiesterase I inhibitors based on an adenosine 5′-(α or γ)-thio-(α, β-or β, γ)-methylenetriphosphate scaffold. J Med Chem 57(11):4677–4691
Lecka J, Ben-David G, Simhaev L, Eliahu S, Oscar J Jr, Luyindula P et al (2013) Nonhydrolyzable ATP analogues as selective inhibitors of human NPP1: a combined computational/experimental study. J Med Chem 56(21):8308–8320
Ahmad H, Ullah S, Rahman F, Saeed A, Pelletier J, Sévigny J et al (2020) Synthesis of biphenyl oxazole derivatives via Suzuki coupling and biological evaluations as nucleotide pyrophosphatase/phosphodiesterase-1 and-3 inhibitors. Eur J Med Chem 208:112759
Anbar HS, El-Gamal R, Ullah S, Zaraei S-O, Al-Rashida M, Zaib S et al (2020) Evaluation of sulfonate and sulfamate derivatives possessing benzofuran or benzothiophene nucleus as inhibitors of nucleotide pyrophosphatases/phosphodiesterases and anticancer agents. Bioorg Chem 104:104305
El-Gamal MI, Ullah S, Zaraei S-O, Jalil S, Zaib S, Zaher DM et al (2019) Synthesis, biological evaluation, and docking studies of new raloxifene sulfonate or sulfamate derivatives as inhibitors of nucleotide pyrophosphatase/phosphodiesterase. Eur J Med Chem 181:111560
Semreen MH, El-Gamal MI, Ullah S, Jalil S, Zaib S, Anbar HS et al (2019) Synthesis, biological evaluation, and molecular docking study of sulfonate derivatives as nucleotide pyrophosphatase/phosphodiesterase (NPP) inhibitors. Bioorg Med Chem 27(13):2741–2752
Ullah S, Pelletier J, Sévigny J, Iqbal J (2022) Synthesis and biological evaluation of arylamide sulphonate derivatives as ectonucleotide pyrophosphatase/phosphodiesterase-1 and-3 inhibitors. ACS Omega 7(30):26905–26918
Patel SD, Habeski WM, Cheng AC, de la Cruz E, Loh C, Kablaoui NM (2009) Quinazolin-4-piperidin-4-methyl sulfamide PC-1 inhibitors: alleviating hERG interactions through structure based design. Bioorg Med Chem Lett 19(12):3339–3343
Jung JE, Jang Y, Jeong HJ, Kim SJ, Park K, Yu A et al (2022) Discovery of 3, 4-dihydropyrimido [4, 5-d] pyrimidin-2 (1H)-one and 3, 4-dihydropyrido [2, 3-d] pyrimidin-2 (1H)-one derivatives as novel ENPP1 inhibitors. Bioorg Med Chem Lett 75:128947
Kuhrt D, Ejaz SA, Afzal S, Khan SU, Lecka J, Sévigny J et al (2017) Chemoselective synthesis and biological evaluation of arylated 2-(Trifluoromethyl) quinolines as nucleotide pyrophosphatase (NPPs) inhibitors. Eur J Med Chem 138:816–829
Ausekle E, Ejaz SA, Khan SU, Ehlers P, Villinger A, Lecka J et al (2016) New one-pot synthesis of N-fused isoquinoline derivatives by palladium-catalyzed C-H arylation: potent inhibitors of nucleotide pyrophosphatase-1 and-3. Org Biomol Chem 14(48):11402–11414
Ullah S, El-Gamal MI, El-Gamal R, Pelletier J, Sevigny J, Shehata MK et al (2021) Synthesis, biological evaluation, and docking studies of novel pyrrolo [2, 3-b] pyridine derivatives as both ectonucleotide pyrophosphatase/phosphodiesterase inhibitors and antiproliferative agents. Eur J Med Chem 217:113339
Choudhary MI, Fatima N, Khan KM, Jalil S, Iqbal S (2006) New biscoumarin derivatives-cytotoxicity and enzyme inhibitory activities. Bioorg Med Chem 14(23):8066–8072
Khan KM, Fatima N, Rasheed M, Jalil S, Ambreen N, Perveen S et al (2009) 1, 3, 4-Oxadiazole-2 (3H)-thione and its analogues: a new class of non-competitive nucleotide pyrophosphatases/phosphodiesterases 1 inhibitors. Bioorg Med Chem 17(22):7816–7822
Khan KM, Siddiqui S, Saleem M, Taha M, Saad SM, Perveen S et al (2014) Synthesis of triazole Schiff bases: novel inhibitors of nucleotide pyrophosphatase/phosphodiesterase-1. Bioorg Med Chem 22(22):6509–6514
Lee S-Y, Perotti A, De Jonghe S, Herdewijn P, Hanck T, Müller CE (2016) Thiazolo [3, 2-a] benzimidazol-3 (2H)-one derivatives: structure–activity relationships of selective nucleotide pyrophosphatase/phosphodiesterase1 (NPP1) inhibitors. Bioorg Med Chem 24(14):3157–3165
Jeong HJ, Lee HL, Kim SJ, Jeong JH, Ji SH, Kim HB et al (2022) Identification of novel pyrrolopyrimidine and pyrrolopyridine derivatives as potent ENPP1 inhibitors. J Enzyme Inhib Med Chem 37(1):2434–2451
Supe L, Afzal S, Mahmood A, Ejaz SA, Hein M, Iaroshenko VO et al (2018) Deazapurine analogues bearing a 1H-pyrazolo [3, 4-b] pyridin-3 (2H)-one core: synthesis and biological activity. Eur J Org Chem 2018(20–21):2629–2644
Jafari B, Yelibayeva N, Ospanov M, Ejaz SA, Afzal S, Khan SU et al (2016) Synthesis of 2-arylated thiadiazolopyrimidones by Suzuki-Miyaura cross-coupling: a new class of nucleotide pyrophosphatase (NPPs) inhibitors. RSC Adv 6(109):107556–107571
Arif M, Shabir G, Ejaz S, Saeed A, Khan S, Lecka J et al (2022) Diacylhydrazine derivatives of 2-(5-(pyridin-3-yl)-2 H-tetrazol-2-yl) acetohydrazide and 2-(5-(pyridin-4-yl)-2 H-tetrazol-2-yl) acetohydrazide as potential inhibitors of nucleotide pyrophosphatase. Russ J Bioorg Chem 48(5):990–1001
Gangar M, Goyal S, Raykar D, Khurana P, Martis AM, Goswami A et al (2022) Design, synthesis and biological evaluation studies of novel small molecule ENPP1 inhibitors for cancer immunotherapy. Bioorg Chem 119:105549
Chang L, Lee S-Y, Leonczak P, Rozenski J, De Jonghe S, Hanck T et al (2014) Imidazopyridine-and purine-thioacetamide derivatives: potent inhibitors of nucleotide pyrophosphatase/phosphodiesterase 1 (NPP1). J Med Chem 57(23):10080–10100
Mihajlovic K, Bukvic MA, Dragic M, Scortichini M, Jacobson KA, Nedeljkovic N (2023) Anti-inflammatory potency of novel ecto-5′-nucleotidase/CD73 inhibitors in astrocyte culture model of neuroinflammation. Eur J Pharmacol 956:175943
Bowman CE, da Silva RG, Pham A, Young SW (2019) An exceptionally potent inhibitor of human CD73. Biochemistry 58(31):3331–3334
Lawson KV, Kalisiak J, Lindsey EA, Newcomb ET, Leleti MR, Debien L et al (2020) Discovery of AB680: a potent and selective inhibitor of CD73. J Med Chem 63(20):11448–11468
Du X, Moore J, Blank BR, Eksterowicz J, Sutimantanapi D, Yuen N et al (2020) Orally bioavailable small-molecule CD73 inhibitor (OP-5244) reverses immunosuppression through blockade of adenosine production. J Med Chem 63(18):10433–10459
Sharif EU, Kalisiak J, Lawson KV, Miles DH, Newcomb E, Lindsey EA et al (2021) Discovery of potent and selective methylenephosphonic acid CD73 inhibitors. J Med Chem 64(1):845–860
Bhattarai S, Freundlieb M, Pippel J, Meyer A, Abdelrahman A, Fiene A et al (2015) α, β-Methylene-ADP (AOPCP) derivatives and analogues: development of potent and selective ecto-5′-nucleotidase (CD73) inhibitors. J Med Chem 58(15):6248–6263
Junker A, Renn C, Dobelmann C, Namasivayam V, Jain S, Losenkova K et al (2019) Structure–activity relationship of purine and pyrimidine nucleotides as ecto-5′-nucleotidase (CD73) inhibitors. J Med Chem 62(7):3677–3695
Bhattarai S, Pippel J, Scaletti E, Idris R, Freundlieb M, Rolshoven G et al (2020) 2-Substituted α, β-methylene-ADP derivatives: potent competitive ecto-5′-nucleotidase (CD73) inhibitors with variable binding modes. J Med Chem 63(6):2941–2957
Bhattarai S, Pippel J, Meyer A, Freundlieb M, Schmies C, Abdelrahman A et al (2019) X-ray co-crystal structure guides the way to subnanomolar competitive ecto-5′-nucleotidase (CD73) inhibitors for cancer immunotherapy. Adv Ther 2(10):1900075
Ghoteimi R, Nguyen VT, Rahimova R, Grosjean F, Cros-Perrial E, Uttaro JP et al (2019) Synthesis of substituted 5′-aminoadenosine derivatives and evaluation of their inhibitory potential toward CD73. ChemMedChem 14(15):1431–1443
Liu S, Li D, Liu J, Wang H, Horecny I, Shen R, et al. 2021 A novel CD73 inhibitor SHR170008 suppresses adenosine in tumor and enhances anti-tumor activity with PD-1 blockade in a mouse model of breast cancer. OncoTargets Ther 4561–74
Wen J, Zhang H, Meng C, Zhou D, Chen G, Wang J, et al. 2021 Computational investigation of adenosine 5′-(α, β-methylene)-diphosphate (AMPCP) derivatives as ecto-5′-nucleotidase (CD73) inhibitors by using 3D-QSAR, molecular docking, and molecular dynamics simulations. Struct Chem 1–2
Ghoteimi R, Braka A, Rodriguez C, Cros-Perrial E, Uttaro J-P, Mathé C et al (2021) 4-Substituted-1, 2, 3-triazolo nucleotide analogues as CD73 inhibitors, their synthesis, in vitro screening, kinetic and in silico studies. Bioorg Chem 107:104577
Channar PA, Bano S, Hassan S, Perveen F, Saeed A, Mahesar PA et al (2022) Appraisal of novel azomethine–thioxoimidazolidinone conjugates as ecto-5′-nucleotidase inhibitors: synthesis and molecular docking studies. RSC Adv 12(27):17596–17606
Grosjean F, Cros-Perrial E, Braka A, Uttaro JP, Chaloin L, Jordheim LP et al (2022) Synthesis and studies of potential inhibitors of CD73 based on a triazole scaffold. Eur J Org Chem 2022(21):e202101175
Beatty JW, Lindsey EA, Thomas-Tran R, Debien L, Mandal D, Jeffrey JL et al (2020) Discovery of potent and selective non-nucleotide small molecule inhibitors of CD73. J Med Chem 63(8):3935–3955
Hassan S, Channar PA, Larik FA, Saeed A, Shah HS, Lecka J et al (2018) Synthesis of novel (E)-1-(2-(2-(4 (dimethylamino) benzylidene) hydrazinyl)-4-methylthiazol-5-yl) ethanone derivatives as ecto-5′-nucleotidase inhibitors. R Soc Open Sci 5(9):180837
Iqbal J, Saeed A, Raza R, Matin A, Hameed A, Furtmann N et al (2013) Identification of sulfonic acids as efficient ecto-5′-nucleotidase inhibitors. Eur J Med Chem 70:685–691
Raza R, Saeed A, Lecka J, Sevigny J, Iqbal J (2012) Identification of small molecule sulfonic acids as ecto-5’-nucleotidase inhibitors. Med Chem 8(6):1133–1139
Lyu S, Zhao Y, Zeng X, Chen X, Meng Q, Ding Z et al (2021) Identification of phelligridin-based compounds as novel human CD73 inhibitors. J Chem Inf Model 61(3):1275–1286
Viviani LG, Piccirillo E, Ulrich H, AT-d, Amaral (2019) Virtual screening approach for the identification of hydroxamic acids as novel human ecto-5′-nucleotidase inhibitors. J Chem Inf Model 60(2):621–30
Ashraf A, Shafiq Z, Khan Jadoon MS, Tahir MN, Pelletier J, Sevigny J et al (2020) Synthesis, characterization, and in silico studies of novel spirooxindole derivatives as ecto-5′-nucleotidase inhibitors. ACS Med Chem Lett 11(12):2397–2405
Rivera RP, Hassan S, Ehlers P, Lecka J, Sévigny J, Rodríguez ET et al (2018) Chemoselective synthesis and human ecto-5′-nucleotidase inhibitory activity of 2-trifluoromethyl-4, 6-diarylquinolines. ChemistrySelect 3(30):8587–8592
Miliutina M, Janke J, Chirkina E, Hassan S, Ejaz SA, Khan SU et al (2017) Domino reactions of chromone-3-carboxylic acids with aminoheterocycles: synthesis of heteroannulated pyrido [2, 3-c] coumarins and their optical and biological activity. Eur J Org Chem 2017(47):7148–7159
Ripphausen P, Freundlieb M, Brunschweiger A, Zimmermann H, Müller CE (2012) Virtual screening identifies novel sulfonamide inhibitors of ecto-5′-nucleotidase. J Med Chem 55(14):6576–81
Baqi Y, Lee S-Y, Iqbal J, Ripphausen P, Lehr A, Scheiff AB et al (2010) Development of potent and selective inhibitors of ecto-5′-nucleotidase based on an anthraquinone scaffold. J Med Chem 53(5):2076–2086
Miliutina M, Janke J, Hassan S, Zaib S, Iqbal J, Lecka J et al (2018) A domino reaction of 3-chlorochromones with aminoheterocycles. Synthesis of pyrazolopyridines and benzofuropyridines and their optical and ecto-5′-nucleotidase inhibitory effects. Org Biomol Chem 16(5):717–32
Warren MC, Matissek S, Rausch M, Panduro M, Hall RJ, Dulak A et al (2023) SRF617 is a potent inhibitor of CD39 with immunomodulatory and antitumor properties. ImmunoHorizons 7(5):366–379
Zhang Y, Hu J, Ji K, Jiang S, Dong Y, Sun L, et al. 2023 CD39 inhibition and VISTA blockade may overcome radiotherapy resistance by targeting exhausted CD8+ T cells and immunosuppressive myeloid cells. Cell Rep Med 4 8
Xu Z, Gu C, Yao X, Guo W, Wang H, Lin T et al (2020) CD73 promotes tumor metastasis by modulating RICS/RhoA signaling and EMT in gastric cancer. Cell Death Dis 11(3):202
Lévesque S, Lavoie ÉG, Lecka J, Bigonnesse F, Sévigny J (2007) Specificity of the ecto-ATPase inhibitor ARL 67156 on human and mouse ectonucleotidases. Br J Pharmacol 152(1):141–150
Lecka J, Gillerman I, Fausther M, Salem M, Munkonda MN, Brosseau JP et al (2013) 8-BuS-ATP derivatives as specific NTPD ase1 inhibitors. Br J Pharmacol 169(1):179–196
Gendron F-P, Halbfinger E, Fischer B, Duval M, D’Orléans-Juste P, Beaudoin AR (2000) Novel inhibitors of nucleoside triphosphate diphosphohydrolases: chemical synthesis and biochemical and pharmacological characterizations. J Med Chem 43(11):2239–2247
Gillerman I, Lecka J, Simhaev L, Munkonda MN, Fausther M, Martín-Satué M et al (2014) 2-Hexylthio-β, γ-CH2-ATP is an effective and selective NTPDase2 inhibitor. J Med Chem 57(14):5919–5934
Brunschweiger A, Iqbal J, Umbach F, Scheiff AB, Munkonda MN, Sévigny J et al (2008) Selective nucleoside triphosphate diphosphohydrolase-2 (NTPDase2) inhibitors: nucleotide mimetics derived from uridine-5′-carboxamide. J Med Chem 51(15):4518–4528
Zebisch M, Krauss M, Schäfer P, Sträter N (2012) Crystallographic evidence for a domain motion in rat nucleoside triphosphate diphosphohydrolase (NTPDase) 1. J Mol Biol 415(2):288–306
Fiene A, Baqi Y, Lecka J, Sévigny J, Müller CE (2015) Fluorescence polarization immunoassays for monitoring nucleoside triphosphate diphosphohydrolase (NTPDase) activity. Analyst 140(1):140–148
Afzal S, Al-Rashida M, Hameed A, Pelletier J, Sévigny J, Iqbal J (2021) Synthesis, in-vitro evaluation and molecular docking studies of oxoindolin phenylhydrazine carboxamides as potent and selective inhibitors of ectonucleoside triphosphate diphosphohydrolase (NTPDase). Bioorg Chem 112:104957
Afzal S, Al-Rashida M, Hameed A, Pelletier J, Sévigny J, Iqbal J (2020) Functionalized oxoindolin hydrazine carbothioamide derivatives as highly potent inhibitors of nucleoside triphosphate diphosphohydrolases. Front Pharmacol 11:585876
Baqi Y, Rashed M, Schaekel L, Malik EM, Pelletier J, Sévigny J et al (2020) Development of anthraquinone derivatives as ectonucleoside triphosphate diphosphohydrolase (NTPDase) inhibitors with selectivity for NTPDase2 and NTPDase3. Front Pharmacol 11:1282
Zebisch M, Baqi Y, Schäfer P, Müller CE, Sträter N (2014) Crystal structure of NTPDase2 in complex with the sulfoanthraquinone inhibitor PSB-071. J Struct Biol 185(3):336–341
Baqi Y, Weyler S, Iqbal J, Zimmermann H, Müller CE (2009) Structure-activity relationships of anthraquinone derivatives derived from bromaminic acid as inhibitors of ectonucleoside triphosphate diphosphohydrolases (E-NTPDases). Purinergic Signal 5:91–106
Shehata MK, Uzair M, Zaraei SO, Shahin AI, Shah SJ, Ullah S et al (2023) Synthesis, biological evaluation, and molecular modeling studies of a new series of imidazothiazole or imidazooxazole derivatives as inhibitors of ectonucleoside triphosphate diphosphohydrolases (NTPDases). Med Chem Res 32(2):314–325
Murtaza A, Afzal S, Zaman G, Saeed A, Pelletier J, Sévigny J et al (2021) Divergent synthesis and elaboration of structure activity relationship for quinoline derivatives as highly selective NTPDase inhibitor. Bioorg Chem 115:105240
Hayat K, Afzal S, Saeed A, Murtaza A, Rahman SU, Khan KM et al (2019) Investigation of new quinoline derivatives as promising inhibitors of NTPDases: synthesis, SAR analysis and molecular docking studies. Bioorg Chem 87:218–226
Abbas S, Afzal S, Nadeem H, Hussain D, Langer P, Sévigny J et al (2022) Synthesis, characterization and biological evaluation of thiadiazole amide derivatives as nucleoside triphosphate diphosphohydrolases (NTPDases) inhibitors. Bioorg Chem 118:105456
Begum Z, Ullah S, Akram M, Uzair M, Ullah F, Pelletier J et al (2022) Identification of thienopyrimidine glycinates as selective inhibitors for h-NTPDases. Bioorg Chem 129:106196
Müller CE, Iqbal J, Baqi Y, Zimmermann H, Röllich A, Stephan H (2006) Polyoxometalates—a new class of potent ecto-nucleoside triphosphate diphosphohydrolase (NTPDase) inhibitors. Bioorg Med Chem Lett 16(23):5943–5947
Khan KM, Salar U, Afzal S, Wadood A, Taha M, Perveen S et al (2019) Schiff bases of tryptamine as potent inhibitors of nucleoside triphosphate diphosphohydrolases (NTPDases): structure-activity relationship. Bioorg Chem 82:253–266
Lecka J, Fausther M, Künzli B, Sévigny J (2014) Ticlopidine in its prodrug form is a selective inhibitor of human NTPDase1. Mediators Inflamm 2014:547480
Bi C, Schäkel L, Mirza S, Sylvester K, Pelletier J, Lee S-Y et al (2023) Synthesis and structure–activity relationships of ticlopidine derivatives and analogs as inhibitors of ectonucleotidase CD39. Bioorg Chem 135:106460
Zhao Y, Chen X, Ding Z, He C, Gao G, Lyu S et al (2021) Identification of novel CD39 inhibitors based on virtual screening and enzymatic assays. J Chem Inf Model 62(21):5289–5304
Sidique S, Ardecky R, Su Y, Narisawa S, Brown B, Millán JL et al (2009) Design and synthesis of pyrazole derivatives as potent and selective inhibitors of tissue-nonspecific alkaline phosphatase (TNAP). Bioorg Med Chem Lett 19(1):222–225
Andleeb H, Hussain M, Ejaz SA, Sevigny J, Farman M, Yasinzai M et al (2020) Synthesis and computational studies of highly selective inhibitors of human recombinant tissue non-specific alkaline phosphatase (h-TNAP): A therapeutic target against vascular calcification. Bioorg Chem 101:103999
Khurshid A, Saeed A, Ashraf Z, Abbas Q, Hassan M (2021) Understanding the enzymatic inhibition of intestinal alkaline phosphatase by aminophenazone-derived aryl thioureas with aided computational molecular dynamics simulations: synthesis, characterization SAR and kinetic profiling. Mol Divers 25:1701–1715
Hosseini Nasab N, Raza H, Shim RS, Hassan M, Kloczkowski A, Kim SJ (2022) Potent alkaline phosphatase inhibitors, pyrazolo-oxothiazolidines: synthesis, biological evaluation, molecular docking, and kinetic studies. Int J Mol Sci 23(21):13262
Chang L, Mébarek S, Popowycz F, Pellet-Rostaing S, Lemaire M, Buchet R (2011) Synthesis and evaluation of thiophenyl derivatives as inhibitors of alkaline phosphatase. Bioorg Med Chem Lett 21(8):2297–2301
Li L, Chang L, Pellet-Rostaing S, Liger F, Lemaire M, Buchet R et al (2009) Synthesis and evaluation of benzo [b] thiophene derivatives as inhibitors of alkaline phosphatases. Bioorg Med Chem 17(20):7290–7300
Channar PA, Irum H, Mahmood A, Shabir G, Zaib S, Saeed A et al (2019) Design, synthesis and biological evaluation of trinary benzocoumarin-thiazoles-azomethines derivatives as effective and selective inhibitors of alkaline phosphatase. Bioorg Chem 91:103137
Saeed A, Khurshid A, Shabir G, Mahmood A, Zaib S, Iqbal J (2020) An efficient synthetic approach toward a sporadic heterocyclic scaffold: 1, 3-oxathiol-2-ylidenes; alkaline phosphatase inhibition and molecular docking studies. Bioorg Med Chem Lett 30(13):127238
Kumar MR, Manikandan A, Sivakumar A, Dhayabaran VV (2018) An eco-friendly catalytic system for multicomponent, one-pot synthesis of novel spiro-chromeno indoline-triones and their anti-prostate cancer potentials evaluated via alkaline phosphatase inhibition mechanism. Bioorg Chem 81:44–54
Bhatti HA, Khatoon M, Al-Rashida M, Bano H, Iqbal N, Yousuf S et al (2017) Facile dimethyl amino group triggered cyclic sulfonamides synthesis and evaluation as alkaline phosphatase inhibitors. Bioorg Chem 71:10–18
Ejaz SA, Saeed A, Siddique MN, un Nisa Z, Khan S, Lecka J, et al. Synthesis, characterization and biological evaluation of novel chalcone sulfonamide hybrids as potent intestinal alkaline phosphatase inhibitors. Bioorg Chem 2017 70:229-36
Al-Rashida M, Raza R, Abbas G, Shah MS, Kostakis GE, Lecka J et al (2013) Identification of novel chromone based sulfonamides as highly potent and selective inhibitors of alkaline phosphatases. Eur J Med Chem 66:438–49
Al-Rashida M, Ejaz SA, Ali S, Shaukat A, Hamayoun M, Ahmed M et al (2015) Diarylsulfonamides and their bioisosteres as dual inhibitors of alkaline phosphatase and carbonic anhydrase: structure activity relationship and molecular modelling studies. Bioorg Med Chem 23(10):2435–2444
Dahl R, Sergienko EA, Su Y, Mostofi YS, Yang L, Simao AM et al (2009) Discovery and validation of a series of aryl sulfonamides as selective inhibitors of tissue-nonspecific alkaline phosphatase (TNAP). J Med Chem 52(21):6919–6925
Iqbal Z, Ashraf Z, Hassan M, Abbas Q, Jabeen E (2019) Substituted phenyl [(5-benzyl-1, 3, 4-oxadiazol-2-yl) sulfanyl] acetates/acetamides as alkaline phosphatase inhibitors: Synthesis, computational studies, enzyme inhibitory kinetics and DNA binding studies. Bioorg Chem 90:103108
Iqbal Z, Iqbal A, Ashraf Z, Latif M, Hassan M, Nadeem H (2019) Synthesis and docking studies of N-(5-(alkylthio)-1, 3, 4-oxadiazol-2-yl) methyl) benzamide analogues as potential alkaline phosphatase inhibitors. Drug Dev Res 80(5):646–654
Abbasi MA, Nazir M, Ur-Rehman A, Siddiqui SZ, Hassan M, Raza H et al (2019) Bi-heterocyclic benzamides as alkaline phosphatase inhibitors: mechanistic comprehensions through kinetics and computational approaches. Archiv der Pharmazie 352(3):1800278
Mumtaz A, Saeed K, Mahmood A, Zaib S, Saeed A, Pelletier J et al (2020) Bisthioureas of pimelic acid and 4-methylsalicylic acid derivatives as selective inhibitors of tissue-nonspecific alkaline phosphatase (TNAP) and intestinal alkaline phosphatase (IAP): Synthesis and molecular docking studies. Bioorg Chem 101:103996
Saeed A, Saddique G, Channar PA, Larik FA, Abbas Q, Hassan M et al (2018) Synthesis of sulfadiazinyl acyl/aryl thiourea derivatives as calf intestinal alkaline phosphatase inhibitors, pharmacokinetic properties, lead optimization, Lineweaver-Burk plot evaluation and binding analysis. Bioorg Med Chem 26(12):3707–3715
Grodner B, Napiórkowska M. 2017 Characterization and inhibition studies of tissue nonspecific alkaline phosphatase by aminoalkanol derivatives of 1, 7-dimethyl-8, 9-diphenyl-4-azatricyclo [5.2. 1.02, 6] dec-8-ene-3, 5, 10-trione, new competitive and non-competitive inhibitors, by capillary electrophoresis. J Pharm Biomed Anal 143 285-90
Miliutina M, Ejaz SA, Khan SU, Iaroshenko VO, Villinger A, Iqbal J et al (2017) Synthesis, alkaline phosphatase inhibition studies and molecular docking of novel derivatives of 4-quinolones. Eur J Med Chem 126:408–420
Miliutina M, Ivanov A, Ejaz SA, Iqbal J, Villinger A, Iaroshenko VO et al (2015) Diversity oriented synthesis of 6-nitro-and 6-aminoquinolones and their activity as alkaline phosphatase inhibitors. RSC Adv 5(74):60054–60078
Khan I, Shah SJA, Ejaz SA, Ibrar A, Hameed S, Lecka J et al (2015) Investigation of quinoline-4-carboxylic acid as a highly potent scaffold for the development of alkaline phosphatase inhibitors: synthesis, SAR analysis and molecular modelling studies. RSC Adv 5(79):64404–64413
Salar U, Khan KM, Iqbal J, Ejaz SA, Hameed A, Al-Rashida M et al (2017) Coumarin sulfonates: new alkaline phosphatase inhibitors; in vitro and in silico studies. Eur J Med Chem 131:29–47
Iqbal J, El-Gamal MI, Ejaz SA, Lecka J, Sévigny J, Oh C-H (2018) Tricyclic coumarin sulphonate derivatives with alkaline phosphatase inhibitory effects: In vitro and docking studies. J Enzyme Inhib Med Chem 33(1):479–484
Jafari B, Ospanov M, Ejaz SA, Yelibayeva N, Khan SU, Amjad ST et al (2018) 2-Substituted 7-trifluoromethyl-thiadiazolopyrimidones as alkaline phosphatase inhibitors. Synthesis, structure activity relationship and molecular docking study. Eur J Med Chem 144:116–27
Altaf R, Nadeem H, Iqbal MN, Ilyas U, Ashraf Z, Imran M et al (2022) Synthesis, biological evaluation, 2D-QSAR, and molecular simulation studies of dihydropyrimidinone derivatives as alkaline phosphatase Inhibitors. ACS Omega 7(8):7139–7154
Ashraf A, Ejaz SA, Rahman SU, Siddiqui WA, Arshad MN, Lecka J et al (2018) Hybrid compounds from chalcone and 1, 2-benzothiazine pharmacophores as selective inhibitors of alkaline phosphatase isozymes. Eur J Med Chem 159:282–291
Ashraf J, Mughal EU, Alsantali RI, Sadiq A, Jassas RS, Naeem N et al (2021) 2-Benzylidenebenzofuran-3 (2 H)-ones as a new class of alkaline phosphatase inhibitors: synthesis, SAR analysis, enzyme inhibitory kinetics and computational studies. RSC Adv 11(56):35077–35092
Meštrović V, Pavela-Vrančič M (2003) Inhibition of alkaline phosphatase activity by okadaic acid, a protein phosphatase inhibitor. Biochimie 85(7):647–650
Lanier M, Sergienko E, Simão AM, Su Y, Chung T, Millán JL et al (2010) Design and synthesis of selective inhibitors of placental alkaline phosphatase. Bioorg Med Chem 18(2):573–579
Ibrar A, Zaib S, Jabeen F, Iqbal J, Saeed A (2016) Unraveling the alkaline phosphatase inhibition, anticancer, and antileishmanial potential of coumarin–triazolothiadiazine hybrids: design, synthesis, and molecular docking analysis. Arch Pharm 349(7):553–565
Petrosyan A, Ghochikyan TV, Ejaz SA, Mardiyan ZZ, Khan SU, Grigoryan T et al (2017) Synthesis of alkynylated dihydrofuran-2 (3H)-ones as potent and selective inhibitors of tissue non-specific alkaline phosphatase. ChemistrySelect 2(20):5677–5683
Faisal M, Shahid S, Ghumro SA, Saeed A, Larik FA, Shaheen Z et al (2018) DABCO–PEG ionic liquid-based synthesis of acridine analogous and its inhibitory activity on alkaline phosphatase. Synth Commun 48(4):462–472
Khan I, Hanif M, Hussain MT, Khan AA, Aslam MAS, Rama NH et al (2012) Synthesis, acetylcholinesterase and alkaline phosphatase inhibition of some new 1, 2, 4-triazole and 1, 3, 4-thiadiazole derivatives. Aust J Chem 65(10):1413–1419
Channar SA, Channar PA, Saeed A, Alsfouk AA, Ejaz SA, Ujan R et al (2022) Exploring thiazole-linked thioureas using alkaline phosphatase assay, biochemical evaluation, computational analysis and structure–activity relationship (SAR) studies. Med Chem Res 31(10):1792–1802
Saeed A, Javaid M, Shah SJA, Channar PA, Shabir G, Tehzeeb A et al (2022) A zomethine-clubbed thiazoles as human tissue non-specific alkaline phosphatase (h-TNAP) and intestinal alkaline phosphatase (h-IAP) Inhibitors: kinetics and molecular docking studies. Mol Diversity 26(6):3241–3254
Aziz H, Mahmood A, Zaib S, Saeed A, El-Seedi HR, Pelletier J et al (2021) Synthesis, characterization, alkaline phosphatase inhibition assay and molecular modeling studies of 1-benzylidene-2-(4-tert-butylthiazol-2-yl) hydrazines. J Biomol Struct Dyn 39(16):6140–6153
Miliutina M, Ejaz SA, Iaroshenko VO, Villinger A, Iqbal J, Langer P (2016) Synthesis of 3, 3′-carbonyl-bis (chromones) and their activity as mammalian alkaline phosphatase inhibitors. Org Biomol Chem 14(2):495–502
Khan NA, Rashid F, Jadoon MSK, Jalil S, Khan ZA, Orfali R et al (2022) Design, synthesis, and biological evaluation of novel dihydropyridine and pyridine analogs as potent human tissue nonspecific alkaline phosphatase inhibitors with anticancer activity: ROS and DNA damage-induced apoptosis. Molecules 27(19):6235
Mustafa MN, Channar PA, Sarfraz M, Saeed A, Ejaz SA, Aziz M et al (2023) Synthesis, kinetic studies and in-silico investigations of novel quinolinyl-iminothiazolines as alkaline phosphatase inhibitors. J Enzyme Inhib Med Chem 38(1):2163394
Ahmed A, Rehman S-u, Ejaz SA, Saeed A, Ujan R, Channar PA et al (2022) Exploring 2-tetradecanoylimino-3-aryl-4-methyl-1, 3-thiazolines derivatives as alkaline phosphatase inhibitors: biochemical evaluation and computational analysis. Molecules 27(19):6766
Abbasi M, Nazir M, Siddiqui S, Raza H, Zafar A, Shah SA et al (2021) Synthesis, in vitro, and in silico studies of N-(substituted-phenyl)-3-(4-phenyl-1-piperazinyl) propanamides as potent alkaline phosphatase inhibitors. Russ J Bioorg Chem 47:1086–1096
Jeffrey JL, Lawson KV, Powers JP (2020) Targeting metabolism of extracellular nucleotides via inhibition of ectonucleotidases CD73 and CD39. J Med Chem 63(22):13444–13465
Lopez V, Schäkel L, Schuh HM, Schmidt MS, Mirza S, Renn C et al (2021) Sulfated polysaccharides from macroalgae are potent dual inhibitors of human ATP-hydrolyzing ectonucleotidases NPP1 and CD39. Mar Drugs 19(2):51
Schäkel L, Schmies CC, Idris RM, Luo X, Lee S-Y, Lopez V et al (2020) Nucleotide analog ARL67156 as a lead structure for the development of CD39 and dual CD39/CD73 ectonucleotidase inhibitors. Front Pharmacol 11:1294
Younus HA, Hameed A, Mahmood A, Khan MS, Saeed M, Batool F et al (2020) Sulfonylhydrazones: design, synthesis and investigation of ectonucleotidase (ALP & e5′ NT) inhibition activities. Bioorg Chem 100:103827
Younus HA, Saeed M, Mahmood A, Jadoon MSK, Hameed A, Asari A et al (2023) Exploring chromone sulfonamides and sulfonylhydrazones as highly selective ectonucleotidase inhibitors: synthesis, biological evaluation and in silico study. Bioorg Chem 134:106450
Schäkel L, Mirza S, Pietsch M, Lee SY, Keuler T, Sylvester K et al (2021) 2-Substituted thienotetrahydropyridine derivatives: allosteric ectonucleotidase inhibitors. Arch Pharm 354(12):2100300
Ghomashi R, Ghomashi S, Aghaei H, Massah S, Massah AR (2023) Recent Advances in biological active sulfonamide based hybrid compounds part C: multicomponent sulfonamide hybrids. Curr Med Chem 30(37):4181–4255
Hassan S, Ejaz SA, Saeed A, Shehzad M, Khan SU, Lecka J et al (2018) 4-Aminopyridine based amide derivatives as dual inhibitors of tissue non-specific alkaline phosphatase and ecto-5′-nucleotidase with potential anticancer activity. Bioorg Chem 76:237–248
Andleeb H, Hameed S, Ejaz SA, Khan I, Zaib S, Lecka J et al (2019) Probing the high potency of pyrazolyl pyrimidinetriones and thioxopyrimidinediones as selective and efficient non-nucleotide inhibitors of recombinant human ectonucleotidases. Bioorg Chem 88:102893
Saeed A, Ejaz SA, Shehzad M, Hassan S, al-Rashida M, Lecka J, et al. 2016 3-(5-(Benzylideneamino) thiazol-3-yl)-2 H-chromen-2-ones: a new class of alkaline phosphatase and ecto-5′-nucleotidase inhibitors. RSC Adv 6(25):21026-36
Channar PA, Shah SJA, Hassan S, Nisa Zu, Lecka J, Sévigny J et al (2017) Isonicotinohydrazones as inhibitors of alkaline phosphatase and ecto-5′-nucleotidase. Chem Biol Drug Des 89(3):365–70
Abdellatif KR, Bakr RB (2018) New advances in synthesis and clinical aspects of pyrazolo [3, 4-d] pyrimidine scaffolds. Bioorg Chem 78:341–357
Al-Rashida M, Batool G, Sattar A, Ejaz SA, Khan S, Lecka J et al (2016) 2-Alkoxy-3-(sulfonylarylaminomethylene)-chroman-4-ones as potent and selective inhibitors of ectonucleotidases. Eur J Med Chem 115:484–94
Acknowledgements
The authors are grateful to Chemistry Department Quaid-i-Azam University Islamabad.
Author information
Authors and Affiliations
Contributions
Aamer Saeed: conceptualization, supervision, manuscript revision, and finalization.
Huzaifa Sharafat Ali: literature survey, manuscript writing, and structure drawing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Conflict of interest
The authors declare no conflict of interest.
Compliance with ethical standards
The authors declare full compliance of ethical standards and have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sharafat, R.H., Saeed, A. Ectonucleotidase inhibitors: targeting signaling pathways for therapeutic advancement—an in-depth review. Purinergic Signalling (2024). https://doi.org/10.1007/s11302-024-10031-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11302-024-10031-0