
Abstract
Dalbergia cochinchinensis Pierre ex Laness. (Fabaceae) is a commercially important tree in Southeast Asia. Although this species is under legal protections, illegal logging and disorderly developments have reduced its populations, and the conservation of this species is currently of much concern. In this study, we determined nucleotide sequences at six chloroplasts and ten nuclear loci in four populations of D. cochinchinensis in Cambodia, followed by population genetic analyses. The average silent nucleotide diversity over the nuclear loci, excluding one with an exceptionally high value, was 0.0057 in the entire population, and the mean F ST across the nuclear loci between each population pair was between 0.135 and 0.467. Thus, the nucleotide diversity in the studied populations was not low compared with that in other tree species, and the level of population differentiation was high. Neutrality test statistics indicated a recent reduction of population size and a subdivision of the population within this species. The divergence times and migration rates were estimated with a likelihood-based method assuming the isolation with migration model. Based on the results, the three populations split 68,000–138,000 years ago, possibly corresponding to the start of the last glacial period, and the level of gene flow among the populations was very low thereafter. Moreover, after the split, population sizes were reduced considerably. Notably, the nucleotide diversity in an insertion sequence in a noncoding region of nuclear C4H was much higher than the mean nucleotide diversity in silent sites across other nuclear genes, indicating that the region was affected by selection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
To conserve the genetic diversity of a species and/or to understand its evolution, we must know the history of the populations of that species, including splitting and admixture of populations, migrations, and changes in population size, because the current genetic status of a species is shaped by the previous structure of the populations. With recent advances in likelihood-based methodologies, the history of populations can be inferred from DNA data (see, for example, Nielsen and Beaumont 2009). DNA sequence polymorphisms are particularly useful in these approaches because the rates and patterns of nucleotide changes are known to some extent, which are required to calculate likelihoods of the observed genetic variation necessary for inferences.
In this study, we investigated the history of Cambodian populations of the Siamese rosewood, Dalbergia cochinchinensis Pierre ex Laness., in the Fabaceae, a target of the global legume diversity assessment (Yahara et al. 2013), using DNA sequence data of nuclear and chloroplast genes. The genus Dalbergia L. f. includes ca. 250 species of trees, shrubs, and climbers and is widely distributed in tropical and subtropical areas worldwide (Lewis et al. 2005). The genus is entomophilous, occasionally propagates through root suckers, and is regenerated by coppicing. The seedpods are normally flattened for wind dispersal. Because the species are associated with symbiotic nitrogen-fixing bacteria, the plants are useful for soil improvement and reforestation (So 2011). Additionally, good fragrances, beautiful wood grain, and various colors are produced by some species of Dalbergia, particularly D. cochinchinensis, which is famous for the good color of the timber. Furthermore, because the wood is hard, durable, and resistant to termites, D. cochinchinensis is used for premium-grade furniture, musical instruments, and art handicrafts, among other uses, and therefore is commercially important (Soonhuae 1994). D. cochinchinensis is widely distributed in lowland mixed deciduous or dry evergreen forests within the countries of Cambodia, Laos, Thailand, and Vietnam on the Indochina peninsula in Southeast Asia (Niyomdham et al. 1997). Nevertheless, under high pressure of logging for timber, populations of D. cochinchinensis are fragmented into many subpopulations, each composed of only a few individuals, and large trees capable of producing flowers/fruits are rarely seen. Thus, many countries have imposed bans on logging, and the IUCN red list ver. 2.3 qualified D. cochinchinensis as vulnerable (VU) in 1998 (IUCN 2013). However, because the timber and artifacts of this species are sold for high prices, illegal logging continues to rapidly decrease the number of individuals/populations.
Because of these conservation concerns, population genetic analyses of D. cochinchinensis have been conducted using allozyme markers in Thailand (Soonhuae 1994), nucleotide sequences at chloroplast loci in Thailand and Laos (Yooyuen et al. 2012), and random amplified polymorphic DNA (RAPD) and inter simple sequence repeat (ISSR) markers in Vietnam (Hien and Phong 2012). These studies show that substantial genetic variation remains within and among the populations of this species. However, because of the low resolution of those markers and uncertainties in typing some of the markers, inferences on the history of populations of D. cochinchinensis have not yet been conducted.
In this work, we sequenced six chloroplast and ten nuclear genes in a total of 48 individuals sampled from four populations of D. cochinchinensis in three Cambodian provinces. In addition to quantifying the variation between and within populations, we used a likelihood-based method implemented in IMa2 (Hey 2010) to infer the history of these populations, assuming the isolation with migration (IM) model of Hey and Nielsen (2004). The split time of the populations was estimated at 68,000–138,000 years ago, and the size of the ancestral population was estimated to be much larger than that of current populations. Additionally, we found a nuclear locus with a very high level of nucleotide diversity.
Materials and methods
Samples
To infer the history of populations and find candidate loci for selection, we need accurate sequence data. Therefore, although high-throughput genetic analyses using next-generation sequencers (NGSs) have become popular, we still perform Sanger sequencing because we have to detect local singleton mutations with high accuracy.
This, along with occasional necessities for subcloning to determine haplotypes, restricted the total number of alleles sequenced to less than around 100 per locus. In addition, to detect selection using the neutrality statistics, we need 20 or more alleles from each population (Simmonsen et al. 1995). Considering the levels of polymorphism that we observed, we decided to use ten or more individuals from only a few populations for our analysis. Although D. cochinchinensis is distributed outside Cambodia, inferring the history of closely related populations is relevant as a first step for understanding the history of the entire population.
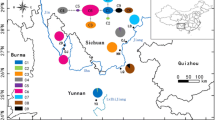
Therefore, leaf samples of D. cochinchinensis were collected in Cambodia at four locations from four populations: one in Siem Reap province (SR), one in Kampong Thom province (KT), and two in Kampot province (KM1 and KM2). Distances between populations were 180 km (SR-KT), 348 km (SR-KM1), 356 km (SR-KM2), 258 km (KT-KM1), 250 km (KT-KM2), and 32 km (KM1-KM2). The sample locations are shown in Fig. 1. Sample information is summarized in Supplementary material Tables S1 and S2. We sampled pieces of leaves from 24 trees in SR, 15 trees in KT, 37 trees in KM1, and 26 trees in KM2. The leaves were dried with silica gel. The sampled trees were separated by at least 3 m. However, some trees shared identical genotypes at all chloroplast and nuclear loci (Supplementary material Table S2). We considered these trees as clones, possibly produced by a root sucker from a nearby tree. Therefore, one of these trees was selected as a representative of the clones in the following analyses. For example, in KM1, leaves from 37 trees were collected at 7 sites (1 to 16 individuals from each site). These sites were more than a hundred meters from one another, but the trees within a site were close to one another. Therefore, all trees at an identical site had the identical genotype. Thus, we selected one tree from each site and used leaves from seven trees for the analyses of KM1. Similarly, 13 trees each were chosen in both KM2 and SR. In KT, all pairs of trees had different genotypes at least at one locus; thus, all 15 trees were used. Because trees were more abundant in KT than in the other three populations, we could sample trees more separately from each other in KT. In total, 48 samples of D. cochinchinensis were used for the analyses. Additionally, four individuals of Dalbergia nigrescens Kurz in Kampong Thom province were sampled and used as out-groups, and one individual of Pterocarpus macrocarpus Kurz, in the identical tribe Dalbergieae, in Siem Reap Province was sampled and used to estimate the nucleotide substitution rate. Total genomic DNA was extracted from dry leaf tissue using the CTAB method (Murray and Thompson 1980).

Locations of sampled populations
Primer design
Universal primers for angiosperms were used for the six chloroplast loci: rbcL (Levin et al. 2003; Kress and Erickson 2007), matK (Dunning and Savolainen 2010), trnS-trnG (Hamilton 1999), trnV-trnM (Demesure et al. 1995), trnLintron (Taberlet et al. 1991), and trnC-petN (Demesure et al. 1995; Lee and Wen 2004). When we could not determine sequences because of long homopolymers, additional sequencing primers were designed to avoid the regions that contained the homopolymers.
PCR primers for the ten nuclear genes were designed based on the sequence information obtained from the NCBI database. Information for the genes is shown in Supplementary material Table S3. Briefly, the primers were designed in the following steps: First, genes were searched with DNA or RNA sequences that were available for more than two species of Fabaceae. The sequences of these genes were aligned, and a pair of primers was designed for conserved regions. When we could amplify the desired segment, the PCR products were sequenced. Then, based on the sequence data, a new primer pair that better matched the gene in Dalbergia was designed. Additionally, internal primers were designed for sequencing as necessary. The primers that were designed are listed in Supplementary material Table S4.
PCR
PCR conditions were an initial 3 min at 95 °C, followed by 35 cycles of 30 s at 95 °C, 30 s at 53 °C, and 1–2 min at 72 °C, and a final 7 min at 72 °C. The products were purified by PEG precipitation treatment to remove surplus primers and dNTPs. The products were directly sequenced on an Applied Biosystems 3730 DNA analyzer using a Big Dye Terminator v3.1 cycle sequence kit (Applied Biosystems, Foster City, CA, USA). Two haplotypes in each individual were determined by subcloning the PCR products using pGEM-T easy vector system I (Promega). All sequence data were assembled using the SeqMan package in Lasergene 10 (DNASTAR, Inc., Madison, WI, USA) and aligned manually using MEGA5 (Tamura et al. 2011).
Data analyses
First, to evaluate the overall genetic structure of the populations, we run STRUCTURE 2.3 (Hubisz et al. 2009) which implements a Bayesian clustering algorithm to find population substructure, with the number of populations, K, set to 1–7. Haplotypes at each locus were regarded as alleles, and the data from all loci were used for the analysis. For each K, ten independent runs, each consisting of 100,000 Markov chain Monte Carlo (MCMC) iterations after a burn-in of 100,000 iterations, were executed. The resulting data were summarized for each K using CLUMPAK (Kopelman et al. 2015), and the mean log-likelihood of the data (Pritchard et al. 2000) and Evanno’s ΔK (Evanno et al. 2005) were calculated to find the optimal value of K using STRUCTURE HARVESTER (Earl and vonHoldt 2012).
The aligned sequences were analyzed using DnaSP v5 (Librado and Rozas 2009). Nucleotide diversity (π; Nei 1987) and haplotype diversity (Hd; Nei 1987) were estimated to assess levels of nucleotide polymorphisms. Neutrality tests based on Tajima’s D (Tajima 1989), Fu and Li’s F and D (Fu and Li 1993), Fay and Wu’s H (Fay and Wu 2000), and Fu’s F s (Fu 1997) were conducted to assess the selective neutrality of mutations. Because we carried out multiple tests in the locus-wise test for each statistic, we evaluated the statistical significance of the results based on the false discovery rate (FDR) of the Benjamini-Hochberg procedure (Benjamini and Hochberg 1995) using the p.adjust function in R version 3.3.2 (R Core Team 2016) for multiple testing. To quantify population differentiation, F ST was estimated using the method of Hudson et al. (1992). Multi-locus tests based on Tajima’s D and Fu and Li’s D were performed using the HKA program (available at https://bio.cst.temple.edu/~hey/software/software.htm#HKA). Haplotype networks were reconstructed using TCS ver. 1.21 (Clement et al. 2000).
Assuming the basic IM model with two populations (Hey and Nielsen 2004), the six parameters (i.e., the divergence time, sizes of the two current and the ancestral populations, and two migration rates for migration in each direction between the two populations) were estimated using the IMa2 program (Hey 2010), which is available at https://bio.cst.temple.edu/~hey/software/software.htm#IMa2. This program calculates likelihoods using coalescent simulation and MCMC simulation. We did not use the data at C4H in this analysis because we suspected that this locus was affected by some type of selection as described later. An estimate of the nucleotide substitution rate per locus per year (U total) is required for the program to estimate the absolute divergence time. To estimate this rate, we used P. macrocarpus, a tree species in the tribe Dalbergieae in the Fabaceae, and sequenced the ten nuclear genes in this species. The estimated divergence time between the Dalbergia species and P. macrocarpus is approximately 49.1 million years ago (mya), according to Lavin et al. (2005), based on a phylogeny estimated by sequences of two cpDNA genes and fossil data of Fabaceae. The total substitution rate per site (K total) between D. cochinchinensis and P. macrocarpus at each nuclear locus was estimated as 0.0895–0.1907 (Supplementary material Table S5) using DnaSP. Let L be the sequence length at each locus and T be the divergence time between the two species; then, U total = (K total × L)/2T. Consequently, U total in D. cochinchinensis at each locus was estimated as 0.46–2.11 × 10−6, with a geometric mean, 1.04 × 10−6, that was used to estimate the divergence time following the instructions described in the IMa2 documentation.
Results
Nucleotide sequences at the six chloroplast and ten nuclear loci were obtained from all 48 samples of D. cochinchinensis, four samples of D. nigrescens, and one sample of P. macrocarpus.
Analysis of chloroplast loci
Only one nucleotide and two mononucleotide repeat polymorphisms were found at the six chloroplast loci (Table 1). The total length sequenced was about 3900 bp. Except in SR, populations were monomorphic but no haplotype was shared between populations, except that between KM1 and KM2, which was an indication that seed migration was rare between populations.
Analysis of nuclear loci
We used STRUCTURE to know the overall genetic structure of the populations, and the result is shown in Supplementary material Fig. S1. The data from all loci were used for this analysis. The optimal number of populations was K = 4 based on the estimated log-likelihood, and K = 3 based on Evanno’s ΔK. When K = 4, except an individual sampled from KM2, individuals from the same population clustered together.
From the analyses using DnaSP, we first noted that nucleotide diversity at C4H was very high, which was caused primarily by very high diversity in a small region 181 or 186 bp in size in the second intron at this locus. Because this region was not found in D. nigrescens, the region was considered an insertion at C4H specific to and fixed in D. cochinchinensis. However, D. nigrescens had an insertion/deletion polymorphism at a different position in the identical intron of C4H, approximately 160 bp upstream of the fixed insertion in D. cochinchinensis. The insertion was 297-bp long and was contained a palindromic structure, with no similarity to the fixed insertion of D. cochinchinensis; therefore, the two insertions in these two species occurred independently.
Population genetic statistics at C4H in D. cochinchinensis are shown in Table 2. Silent nucleotide diversity in the inserted region (π sil = 0.0688) was 9-fold higher than that in the other regions at C4H (π sil = 0.0077) and 12-fold higher than the mean at silent sites across the other nine loci (π sil = 0.0057; Table 3). Fu and Li’s D* and F* were significantly positive in the inserted region. The inserted region was apparently noncoding; however, the sequence was palindromic, and if transcribed, the RNA transcript would assume a secondary structure of a long-stem loop, similar to primary micro RNAs (miRNAs) (Nozawa et al. 2012). The secondary structure was strongly conserved within D. cochinchinensis, although nucleotide diversity was very high in this region. Therefore, compensatory mutations within the inserted region likely occurred to conserve the structure. Because we suspected that the region and therefore C4H were affected by some type of natural selection, C4H was excluded in some of the later analyses including those using IMa2.
Means of diversity measures across the nine nuclear loci, excluding C4H, are summarized in Table 3, and the values at each locus are shown in Supplementary material Table S6. The mean nucleotide diversity at silent sites (π sil) and haplotype diversity (Hd) for the entire population were 0.0057 and 0.740, respectively. The results of the neutrality tests are shown in Table 4 and Supplementary material Table S7. Tajima’s D and Fu and Li’s D and F were mostly positive at the respective loci. Means of Tajima’s D and Fu and Li’s D for the entire population were 0.907 (P = 0.0046) and 0.951 (P = 0.0018), respectively, with both being significantly positive. These results suggested either a population decline, population subdivision, or both.
The mean nucleotide diversity (π sil) within each population ranged from 0.0027 to 0.0050 and was lower than that in the entire population (Table 3). Notably, π sil in KT was half that of the entire population. The haplotype diversity and number of haplotypes in each population were also smaller than those in the entire population. Thus, the genetic variation within each population was lower than that in the entire population. Tajima’s D and Fu and Li’s D and F were positive, with the exception of a few loci (Table 4). Values of Tajima’s D at SQS showed significant excesses of high-frequency polymorphisms in each population of KT and KM2. The values of Fu’s F S at this locus showed that the number of haplotypes was significantly lower than the neutral expectation based on the number of segregating sites in those populations. Tajima’s D was significantly negative at pi3k in KT and KM2. These cases might indicate selective sweeps within each population. The means of Tajima’s D and Fu and Li’s D within each population were all positive and were statistically significant with the exception of Fu and Li’s D in KT, indicating reduction in population size. Fu’s F S was significantly positive at a few loci in addition to SQS in each population. Estimates of F ST are shown in Table 5. The estimate at each locus varied considerably from − 0.048 to 0.926, but the means across loci were generally large, ranging from 0.306 to 0.467, with the exception of 0.135 between KM1 and KM2, which were located near one another. Based on these results, subdivision between populations and reduction in the size of each population were suggested for D. cochinchinensis. The former observation is consistent with the result obtained from the analysis using STRUCTURE.
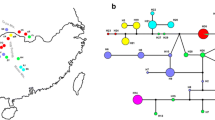
Haplotype networks at the ten nuclear loci in D. cochinchinensis are shown in Fig. 2. The shapes of networks varied considerably among the loci. As expected from the high level of diversity, a complex network was found at C4H. Notably, population differentiation was apparently strong at this locus with many private haplotypes. At CPK, a very distant haplotype was found in SR, whereas no such distant haplotype was found in any population at NCED.

Haplotype networks of genes at each of the ten nuclear loci in D. cochinchinensis. Circles represent haplotypes that are colored according to the sampled locations. The size of each circle represents haplotype frequency. Black dots represent missing haplotypes. Gray two-way arrows in the network of C4H represent ambiguity due to too many possible branches and dots at this locus
History of populations
The divergence time between populations, sizes of the current and ancestral populations, and migration rates, assuming the IM model with two populations, were estimated using the likelihood-based method implemented in IMa2. The two populations in Kampot, KM1 and KM2, were merged in the analysis because the two populations were geographically close to one another, most haplotypes were shared, and F ST between them was relatively low (0.135). The analyses were performed for all pairs of the three populations, with results shown in Table 6 and Fig. 3. Modes of the posterior distributions of divergence times were 68,000 years between KT and SR; 138,000 years between KM and SR; and 113,000 years between KT and KM. The divergence time between KT and SR was the most recent, although the highest posterior density (HPD) intervals overlapped substantially between the population pairs. The population size parameters of the current populations, θ 0 and θ 1, ranged from 0.235 to 0.455. By contrast, the population size parameter, θ A, of the ancestral population in each pair ranged from 2.885 to 3.695, 7- to 13-fold larger than that of current populations. Although the modes of the migration parameters were nonzero, the lower 95% HPDs all included zero, which showed that migration between populations after the splits was weak in all pairs.

Posterior probability densities of a divergence time between and b ancestral population size of each pair of populations estimated by the IMa2 program
Discussion
Selection at some of the nuclear loci
Because populations of D. cochinchinensis apparently experienced recent size reductions as indicated by the positive means of Tajima’s D and Fu and Li’s D and inferred by the IM analysis, any positively significant (here, in the sense of FDR) values of these statistics might not indicate deviations from the neutral expectation. However, Tajima’s D at SQS was positively significant in KT and KM2, and the other populations also showed the same trend. In addition, the mean of F ST values at SQS (0.061) was the lowest among the nine loci (Table 5). This can be visually seen in the haplotype network at the locus. Haplotypes were clustered into two groups, and both groups were represented with intermediate frequencies in each population (Fig. 2). Because the result of the IM analysis suggested that the migration rates between populations were very low, two types of haplotypes might have been maintained within each population by balancing selection after the population split.
Conversely, some statistics with significantly negative values might indicate selective sweeps at the loci because recent size reduction was indicated by the positive mean values of statistics like Tajima’s D. Indeed, Tajima’s D at pi3k in KT and KM2 was significantly negative (Table 4). At this locus in those populations, Fay and Wu’s H was also negative and F ST values were high (Table 5), thereby supporting the occurrences of recent local selective sweeps. Very weak migration between populations after the split might have facilitated the occurrence of local selective sweeps.
Because the secondary structure was conserved among samples in spite of the high level of diversity in this region, the insert in an intron at C4H was most likely a functional noncoding RNA. In Arabidopsis, the corresponding intron has a conserved element near the 5′ end, which suggests some functional importance of this intron (Chen et al. 2007). At present, we do not have any information on the function or high diversity of this intron.
Such noncoding regions with high levels of nucleotide diversity are also reported in other plants. For example, Meaux et al. (2008) found a miRNA, ath-MIR824, with a high level of diversity in Arabidopsis thaliana and suggested that the high nucleotide diversity was caused by balancing selection maintaining structurally different alleles of the miRNA. Because Fu and Li’s D* and F* for the insert at C4H were significantly positive (Table 2) and the secondary structure was conserved, some type of balancing selection might be responsible for the high diversity. The insert is possibly a primary miRNA, but additional experiments and analyses must be conducted to elucidate the biological significance.
Comparison with previous research on D. cochinchinensis
Because of conservation concerns, population genetic analyses of D. cochinchinensis were previously conducted in Thailand, Laos, and Vietnam using different genetic markers. First, Soonhuae (1994) investigated 8 natural populations of D. cochinchinensis in Thailand using 18 allozyme markers, and the estimates of the average heterozygosity and F ST were 0.229 and 0.127, respectively. This author found two population clusters, with one in central and the other in northeastern Thailand. Second, Yooyuen et al. (2012) surveyed DNA sequence variation in three chloroplast intergenic regions in ten natural populations in Thailand and Laos, and the overall nucleotide diversity was estimated as 0.00056. These authors found one primary haplotype was shared by nine of ten populations in Thailand, but the remaining haplotypes were found only in one or two populations, which might suggest intermediate levels of genetic differentiation among current populations caused by fragmentation of an ancient large population. Finally, Hien and Phong (2012) surveyed two populations in Vietnam using RAPD and ISSR markers. These authors estimated an F ST between the two populations, separated by a distance of 300 km, of 0.31; however, this F ST value might be an overestimate because the diversity within one of the populations was close to zero and the sample was suspected to consist of clones.
Although direct comparisons of our results with those three studies were difficult because of the differences in the markers used and/or the populations studied, some common features and differences were noted. First, although D. cochinchinensis is currently fragmented into small populations, the species maintains a fair amount of genetic diversity at nuclear loci; the mean silent diversity at nuclear loci was 0.0057. The mean silent nucleotide diversity in each of the 17 other tree species ranges from 0.00003 to 0.01638, with the mean across the tree species of 0.00745 (Lynch 2006; Iwanaga et al. 2012). Thus, the nucleotide diversity in D. cochinchinensis was at an intermediate level among those tree species, which was consistent with the conclusions reached by Soonhuae (1994) for allozymes.
Second, the estimates of F ST at nuclear loci ranged from 0.127 to 0.467 in our study, and those of the other studies using different markers also show moderate to strong differentiation between populations. This level of differentiation was expected because this species is entomophilic. Moreover, the flat seedpods are wind-dispersed and the size (5–6 cm in length) makes it difficult for seedpods to scatter over a long distance (Niyomdham et al. 1997). These characteristics reduce gene flow between populations and are expected to result in relatively high levels of differentiation. Of note, when F ST was only considered between populations separated by more than 200 km, our estimates based on the nuclear gene sequences in Cambodia were higher than those in Thailand based on allozyme markers (Soonhuae 1994) or in Vietnam based on RAPD and ISSR markers (Hien and Phong 2012). At the chloroplast loci, each population was monomorphic in Cambodia with a population-specific haplotype, whereas some of the populations were polymorphic and most populations shared one dominant haplotype in Thailand (Yooyuen et al. 2012). Therefore, although F ST estimates at nuclear loci obtained by different markers might not be comparable, Cambodian populations of D. cochinchinensis might be more strongly differentiated than those in Thailand. However, good estimates of F ST using nuclear gene sequences are required for other countries to obtain a clear picture of the population differentiation of this species within the entire range.
Population history of D. cochinchinensis
The estimation using IMa2 showed that the Cambodian populations of D. cochinchinensis apparently separated from one another approximately 100,000 years ago (kya), with little gene flow after the split. Additionally, a reduction in population size from that of the ancestral population after the split was inferred from our analysis. The reduction in population size was also supported by the mostly positive values of Tajima’s D in each population. In other words, population fragmentation seemed to have occurred 100 kya in the Cambodian population of D. cochinchinensis and this condition has continued until now. We attempted to relate these histories of the populations to past environmental changes in Indochina as follows.
D. cochinchinensis generally grows in mixed deciduous or dry evergreen forests in which a distinct dry season occurs with less frequent rainfall events from December to March and a rainy season occurs from April to November (Miyazawa et al. 2014). The estimated times, ca. 100 kya, of the splits of the populations correspond to the early ages of the last glacial period that lasted from 110 to 12 kya. Based primarily on palynological studies, the climatic conditions in Indochina were likely generally cooler and more arid from ca. 30 to 11.9 kya than those at present (Cook and Jones 2012). This trend might extend to 110 kya when the last glacial period began, although little information is available on this period because palynological data in the period between 30 and 110 kya is currently scant for this region. Indeed, fossil data of large- and medium-bodied animals indicate that the lands of Indochina in the middle Pleistocene were most likely of heterogeneous habitats consisting of patches of grassland and forests of the dry evergreen type (Louys and Meijaard 2010).
Because suitable habitats for D. cochinchinensis are somewhat arid and the climate was arid throughout the last glacial period, why the splits occurred in the populations at approximately the beginning of the last glacial period was not clear, when we considered the IMa estimates at face value. Additionally, expansions might be expected at that time, not reductions. Although hunter-gatherers were likely in Indochina approximately 43 kya (White et al. 2004), the influence of these people was likely small because they did not rely on agriculture and the hardwood of D. cochinchinensis would be difficult to cut with primitive stone axes. Unfortunately, as mentioned above, palynological data before 40 kya in Indochina is lacking (Penny 2001), making identification of past forest types in respective areas of Indochina difficult. Thus, at present, we cannot specify the exact cause of the splits and the reductions in these populations.
Conclusion
Because we used sequencing rather than genotyping at microsatellite or allozyme loci in this study, we could infer population fragmentation that occurred around 100 kya with very weak gene flow between populations afterwards. Moreover, we could find a few candidate genes for selection. Although we currently cannot identify environmental factors that caused the fragmentation of the population of D. cochinchinensis in Cambodia, a few insights, possibly useful for conservation of this species, could be obtained. Firstly, variation between populations is caused by the lack of gene flow rather than the balance between gene flow and drift. Therefore, maintaining each local population at the location with each locally adapted variation intact is important. Secondly, in addition to a locally adapted locus, a locus affected by balancing selection was found (SQS). Balanced polymorphisms may be lost if population size becomes too small (Kimura and Crow, 1964). Therefore, keeping sizes of local populations large enough is also important. Finally, splits of a large population into smaller populations and the lack of gene flow between them likely lasted throughout the last glacial period, and therefore, the fragmented status of the populations itself is not likely to be of concern for conservation.
However, recent reductions in the sizes of D. cochinchinensis populations are caused by artificial logging. Although the current level of genetic diversity in the total population remains high, because the level of gene flow between populations has been low, as shown in this study and others, and only a few generations have passed since the beginning of artificial logging, losses of genetic diversity would be large if some of the local populations are lost completely. Moreover, in the currently surviving populations from which we obtained samples, the populations consisted primarily of young trees shorter than 15 m and full-grown trees were rare because large trees were logged. Because only full-grown trees reproduce, if such logging of large trees continues, the next generation of those populations will not be produced and the genetic variation that we reported in this study will be lost. Thus, conservation plans must be designed to maintain locally adapted variation by maintaining geographically separated populations, each containing adult trees.
References
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B Methodol 57(1):289–300. https://doi.org/10.2307/2346101
Chen AH, Chai YR, Li JN, Chen L (2007) Molecular cloning of two genes encoding cinnamate 4-hydroxylase (C4H) from oilseed rape (Brassica Napus). J Biochem Mol Biol 40(2):247–260
Clement M, Posada D, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9(10):1657–1659. https://doi.org/10.1046/j.1365-294x.2000.01020.x
Cook GC, Jones RT (2012) Paleoclimate dynamics in continental Southeast Asia over the last ~30,000 Cal yrs BP. Palaeogeogr Palaeoclimatol Palaeoecol 339–341:1–11. https://doi.org/10.1016/j.palaeo.2012.03.025
Core Team R (2016) R: a language and environment for statistical computing. In: R foundation for statistical computing, Vienna. URL https://www.R-project.org/
Demesure B, Sodzi N, Petit RJ (1995) A set of universal primers for amplification of polymorphic non-coding regions of mitochondrial and chloroplast DNA in plants. Mol Ecol 4(1):129–131. https://doi.org/10.1111/j.1365-294X.1995.tb00201.x
Dunning LT, Savolainen V (2010) Broad-scale amplification of matK for DNA barcoding plants, a technical note. Bot J Linn Soc 164(1):1–9. https://doi.org/10.1111/j.1095-8339.2010.01071.x
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4(2):359–361. https://doi.org/10.1007/s12686-011-9548-7
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14(8):2611–2620
Fay JC, Wu CI (2000) Hitchhiking under positive Darwinian selection. Genetics 155(3):1405–1413
Fu YX (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147(2):915–925
Fu YX, Li WH (1993) Statistical tests of neutrality of mutations. Genetics 133(3):693–709
Hamilton MB (1999) Four primer pairs for the amplification of chloroplast intergenic regions with intraspecific variation. Mol Ecol 8(3):521–523. https://doi.org/10.1046/j.1365-294x.1999.00510.x
Hey J (2010) Isolation with migration models for more than two populations. Mol Biol Evol 27:905–920
Hey J, Nielsen R (2004) Multilocus methods for estimating populations sizes, migration rates and divergence time, with application to the divergence of Drosophila pseudooscura and D. persimillis. Genetics 167:747–760
Hien VTT, Phong DT (2012) Genetic diversity among endangered rare Dalbergia cochinchinensis (Fabaceae) genotypes in Vietnam revealed by random amplified polymorphic DNA (RAPD) and inter simple sequence repeats (ISSR) markers. Afr J Biotechnol 11(35):8632–8644. https://doi.org/10.5897/AJB11.3598
Hubisz MJ, Falush D, Stephens M, Pritchard JK (2009) Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour 9:1322–1332. https://doi.org/10.1111/j.1755-0998.2009.02591.x
Hudson RR, Slatkin M, Maddison WP (1992) Estimation of levels of gene flow from DNA sequence data. Genetics 132:583–589
Iwanaga H, Teshima KM, Khatab IA, Inomata N, Finkeldey R, Siregar IZ, Szmidt AE (2012) Population structure and demographic history of a tropical lowl and rainforest tree species Shorea parvifolia (Dipterocarpaceae) from southeastern Asia. Ecol Evol 2(7):1663–1675. https://doi.org/10.1002/ece3.284
Kimura M, Crow JF (1964) The number of alleles that can be maintained in a finite population. Genetics 49:725–738
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) CLUMPAK: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15(5):1179–1191. https://doi.org/10.1111/1755-0998.12387
Kress WJ, Erickson DL (2007) A two-locus global DNA barcode for land plants: the coding rbcL gene complements the non-coding trnH-psbA spacer region. PLoS One 2(6):e508. https://doi.org/10.1371/journal.pone.0000508
Lavin M, Herendeen PS, Wojciechowski MF (2005) Evolutionary rates analysis of Leguminosae implicates a rapid diversification of lineages during the tertiary. Syst Biol 54(4):575–594. https://doi.org/10.1080/10635150590947131
Lee C, Wen J (2004) Phylogeny of panax using chloroplast trnC-trnD intergenicregion and the utility of trnC-trnD in interspecific studies of plants. Mol Phylogenet Evol 31(3):894–903. https://doi.org/10.1016/j.ympev.2003.10.009
Levin RA, Wagner WL, Hoch PC, Nepokroeff M, Pires JC, Zimmer EA, Sytsma KJ (2003) Family-level relationships of Onagraceae based on chloroplast rbcL and ndhF data. Am J Bot 90(1):107–115. https://doi.org/10.3732/ajb.90.1.107
Lewis G, Schrire B, Mackinder B, Lock M (2005) Legumes of the world. Royal Botanic Gardens, Kew
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25(11):1451–1452. https://doi.org/10.1093/bioinformatics/btp187; 10.1093/bioinformatics/btp187
Louys J, Meijaard E (2010) Palaeoecology of southeast Asian megafauna-bearing sites from the Pleistocene and a review of environmental changes in the region. J Biogeogr 37(8):1432–1449. https://doi.org/10.1111/j.1365-2699.2010.02297.x
Lynch M (2006) The origins of eukaryotic gene structure. Mol Biol Evol 23(2):450–468. https://doi.org/10.1093/molbev/msj050
Meaux J, JY H, Tartler U, Goebel U (2008) Structurally different alleles of the ath-MIR824 microRNA precursor are maintained at high-frequency in Arabidopsis thaliana. Proc Natl Acad Sci U S A 105(26):8994–8999. https://doi.org/10.1073/pnas.0803218105
Miyazawa Y, Tateishi M, Komatsu H, Ma V, Kajisa T, Sokh H, Mizoue N, Kumagai T (2014) Tropical tree water use under seasonal waterlogging and drought in central Cambodia. J Hydrol 515:81–89. https://doi.org/10.1016/j.jhydrol.2014.04.049
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8(19):4321–4325. https://doi.org/10.1093/nar/8.19.4321
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Nielsen R, Beaumont MA (2009) Statistical inferences in phylogeogrphy. Mol Ecol 18:2034–1047
Niyomdham C, Pham Hoang Hô, Dy Phon P, Vidal JE (1997) Flore du Cambodge du Laos et du Vietnam vol. 29, Leguminoseae-Papilionoïdeae Dalbergieae. Paris: Museum National d'Histoire Naturelle
Nozawa M, Miura S, Nei M (2012) Origins and evolution of microRNA genes in plant species. Genome Biol Evol 4(3):230–239. https://doi.org/10.1093/gbe/evs002
Penny D (2001) A 40,000 year palynological record from north-east Thailand: implications for biogeography and paleo-environmental reconstruction. Palaeogeogr Palaeoclimatol Palaeoecol 171:97–128
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Simonsen KL, Churchill GA, Aquadro CF (1995) Properties of statistical tests of neutrality for DNA polymorphism data. Genetics 141:413–429
So T (2011) Improving reforestation success of high-value and key forest species by direct seeding in Southeast Asia and Western Australia. School of Biological Sciences and Biotechnology, Faculty of Science, Engineering and Sustainability, Murdoch University. Thesis
Soonhuae P (1994) Estimation of genetic variation in Thailand rosewood (Dalbergia cochinchinensis Pierre). Kasetsart University, Thailand. Thesis. doi:10.14288/1.0099229Taberlet
Taberlet P, Gielly L, Pautou G, Bouvet J (1991) Universal primers for amplification of three non-coding regions of chloroplast DNA. Plant Mol Biol 17(5):1105–1109. https://doi.org/10.1007/BF00037152
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123(3):585–595
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28(10):2731–2739. https://doi.org/10.1093/molbev/msr121; 10.1093/molbev/msr121
The IUCN red list of threatened species. Version 2013.2. http://www.iucnredlist.org/
White JC, Penny D, Kealhoefer L, Maloney B (2004) Vegetation changes from the late Pleistocene through the Holocene from three areas of archaeology significance in Thailand. Quat Int 113:111–132
Yahara T, Javadi F, Onoda Y et al (2013) Global legume diversity assessment: concepts, key indicators and strategies. Taxon 62:249–266. 10.12705/622.12
Yooyuen R, Duangjai S, Changtragoon S (2012) Chloroplast DNA phylogeography of Dalbergia cochinchinensis Pierre in Thailand and Laos. (ISSN 1016-3263, ISBN 978-3-902762-15-3)
Acknowledgements
We cordially thank the Forestry Administration of Cambodia for the permits and support of our fieldwork in Cambodia.
Data archiving statement
All sequences are deposited with EMBL/GenBank/DDBJ under the accession numbers AB850618–AB850637 for the chloroplast loci and KX428531–KX429590 for the nuclear loci.
Funding
The Environment Research and Technology Development Fund (S9) of the Ministry of Environment, Japan, and JSPS KAKENHI grant numbers JP26291082 and JP16H02553 supported this research.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by D. B. Neale
Electronic supplementary material
ESM 1
(PDF 516 kb)
Rights and permissions
About this article

Cite this article
Moritsuka, E., Chhang, P., Tagane, S. et al. Genetic variation and population structure of a threatened timber tree Dalbergia cochinchinensis in Cambodia. Tree Genetics & Genomes 13, 115 (2017). https://doi.org/10.1007/s11295-017-1199-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-017-1199-8