
Abstract
Historical evolutionary events highly affect the modern-day genetic structure of natural populations. Scots pine (Pinus sylvestris L.), as a dominant tree species of the Eurasian taiga communities following the glacial cycles of the Pleistocene, has survived in small, scattered populations at the range limits of its south-eastern European distribution. In this study, we examined genetic relationships, genetic divergence and demographic history of peripheral populations from central-eastern Europe, the Carpathian Mountains and the Pannonian Basin. Four hundred twenty-one individuals from 20 populations were sampled and characterized with both nuclear and chloroplast simple sequence repeat (SSR) markers. Standard population genetic indices, the degree of genetic differentiation and spatial genetic structure were analysed. Our results revealed that peripheral Scots pine populations retained high genetic diversity despite the recently ongoing fragmentation and isolation of the persisting relict populations. Analysis of molecular variance (AMOVA) showed 7% among-population genetic differentiation, and there was no isolation by distance among the island-like occurrences. Genetic discontinuities with strong barriers (99–100% bootstrap support) were identified in the Carpathians. Based on both marker types, populations of the Western Carpathians were delimited from those inhabiting the Eastern Carpathians, and two main genetic lineages were traced that most probably originate from two main refugia. One refugium presumably existed in the region of the Eastern Alps with the Hungarian Plain, while the other was probably found in the Eastern Carpathians. These findings are supported by recent palynological records. The strongest genetic structure was revealed within the Romanian Carpathians on the basis of both marker types. With only some exceptions, no signs of recent bottlenecks or inbreeding were detected. However, Carpathian natural populations of Scots pine are highly fragmented and have a small census size, though they have not yet been affected by genetic erosion induced by isolation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Among various factors, landscape-scale habitat loss, population fragmentation and isolation impose the highest risks for species survival (Fahrig 2003; Ewers and Didham 2006). The long-lasting effects of fragmentation, which also trigger a loss of genetic diversity, may ultimately lead to population breakdown and extinction (Chapin et al. 2000). Natural (climate-driven) changes on a long-term historical timescale can leave imprints in spatial genetic patterns and the demography of a species’ current spatial genetic structure (Bradshaw 2004). However, in the case of forest trees, genetic signals caused by fragmentation, such as inbreeding, genetic isolation and bottlenecks, appear in the population structure over a much longer period of time due to their long generation time and the effects of slow mutation, nucleotide substitution and speciation rates (Savolainen and Pyhäjärvi 2007; Kramer et al. 2008). This period can be as long as hundreds or thousands of years in conifer species (Petit and Hampe 2006). Peripheral populations living at the margin of a species’ natural range are likely vulnerable to the genetic effects of population fragmentation (Lönn and Prentice 2002). Furthermore, isolation on the periphery potentially exposes populations to maladaptation, which can be caused by drift, inbreeding, selection, pleiotropy, linkage disequilibrium, homozygote disadvantage and gene flow (Crespi 2000; Kremer et al. 2012). On a spatial scale, these peripheral populations reflect the species’ physiological boundaries, and they may also represent the climatic and adaptive limits of dispersal (Sykes et al. 1996). Therefore, peripheral populations are important for the future evolution and adaptation of a species, and they can serve as an initial gene pool for speciation (Lesica and Allendorf 1995; Chhatre and Rajora 2014). Thus, peripheral populations are considered important sources in phylogeographic, population genetic and demographic studies.
Scots pine (Pinus sylvestris L.) is a long-lived coniferous tree species of the Pinaceae family (Pravdin 1969), which occupies a continuous range as the dominant tree species of the Eurasian taiga communities. Furthermore, it is an essential species of various forest ecosystems (Matías and Jump 2012; Giertych and Mátyás 2013).
Peripheral populations of P. sylvestris have been widely studied across species’ distribution range. Enzymatic polymorphism revealed overall low structuring of populations, but elevated differentiation was reported between populations that derived from different glacial refugia (Müller-Starck et al. 1992). Evaluations involving stands from the southern European provenances showed that they were distinct from the northern European populations (Mejnartowicz 1979; Kieliszewska-Rokicka 1981). Modern-day organelle and nuclear DNA marker studies highlighted that populations, especially on the southern margin of the Mediterranean region, present high genetic diversity and a complex geographic pattern (Robledo-Arnuncio et al. 2005). Similarly, genetic differentiation and high genetic diversity were identified in geographic regions of the Alps and the Apennines, at the species’ Italian periphery (Labra et al. 2006; Scalfi et al. 2009; Belletti et al. 2012). The westernmost populations in the British Isles share a similar pattern, as the Scottish populations differ from the populations in continental Europe (Provan et al. 1998; Wachowiak et al. 2011). On a regional scale, populations from central-eastern Europe, and particularly the Carpathian Mountains and the Pannonian Basin, were recently studied by Bernhardsson et al. (2016). They revealed a low level of differentiation among the populations and the impact of Holocene population fragmentation.
Scots pine along the Carpathian Mountain range is distributed in island-like isolated populations (Fekete and Blattny 1913), but there are also scattered natural populations sustained in mixed forest stands, with broad-leaved species in the western Pannonian Basin at the foothills of the Alps (Pócs 1960; Fekete et al. 2014). The genetic structure of these peripheral populations of the Carpathian distribution was highly affected by the postglacial climate warming, forcing Scots pine to immigrate into edaphically specialized habitat types. Indeed, Scots pine natural populations are distributed in the Carpathians on a large elevation gradient, located in sites of divergent ecological conditions, including humid, cool peatbogs and sunny, dry, rocky outcrops. In addition, historical human-mediated activities further increased habitat fragmentation and considerably reduced population census sizes. In part as a consequence of this, only isolated and island-like populations have been sustained (Giertych and Mátyás 2013).
Drawing on macrofossil and pollen evidence, studies on the Quaternary vegetation history of P. sylvestris within this region conclude that Scots pine, along with other cold-tolerant and drought-tolerant conifer taxa, inhabited the Carpathians and the Pannonian Basin in the full glacial and later in the beginning of the postglacial period (Rudner et al. 1995; Rudner and Sümegi 2001; Jankovská and Pokorný 2008). In situ findings also suggest that conifers and in particular boreal and cool temperate tree taxa like Scots pine in central Europe and in the Carpathians survived the last glacial maximum (LGM 20,000–19,000 Before Present) in small, patchy and discontinuous glacial refugia (Willis et al. 1998; Magyari et al. 2014). Species would have had sustained populations in isolated, so-called cryptic refugia, with favourable conditions both for Scots pine and for other boreal and temperate species (Rull 2009, 2010; Sommer and Zachos 2009). Altogether, Scots pine had a complex spatio-temporal history in central-eastern Europe during the Holocene, influenced mainly by oscillations in the climate and also by anthropogenic impact, as a consequence of which overall reduction of population size was experienced (Feurdean et al. 2007). Pollen records also indicate that populations on the lowlands of the Pannonian Basin dramatically declined during the Holocene (Willis et al. 1995; Magyari 2011). Furthermore, on mid-altitudinal to high-altitudinal sites in the Carpathians, species abundance varied greatly by location (Willis 1994; Birks and Ammann 2000).
Despite combined pollen, macrofossils and organelle DNA analysis that could detect glacial refugia in the Carpathian Basin along the Danube, previous molecular studies performed in the region reported a lack of geographic structure both with mtDNA and cpDNA within the Carpathian Mountains (Cheddadi et al. 2006; Bernhardsson et al. 2016). Similarly, no variation and no phylogeographic structure in mitochondrial DNA were found in provenance trials conducted in the region by Čelepirović et al. (2009).
In this study, we combined nuclear and chloroplast microsatellite markers to discuss the results in the light of putative refugia occurring within the Carpathian region and to (1) highlight the current population structure of the selected peripheral populations from central-eastern Europe, most of which formerly were not included in molecular studies; (2) identify genetic relationships, degree of diversity and degree of divergence and infer gene flow between the stands; and (3) circumscribe putative refugia that existed in the time of the Pleistocene.
Material and methods
Plant material
Altogether, 20 natural and autochthonous populations were collected from the highly fragmented distribution of the species. In total, 421 individuals were analysed with nuclear and chloroplast SSRs (Table 1): five populations from the Pannonian Basin in western Hungary (HFE, HKO, HOR, HZA, HVA); four from the Western Carpathians (Tatra Mountains), i.e. the Low Tatras (STU, SLI) and the westernmost ridges of the High Tatras (SKV and SME); nine from Romania, four of which are found in the Transylvanian Central-Island Mountains (Apuseni) (RBI, RBE, RML, RMH); one in the western part of the Southern Carpathians, Latoriței (RPA); and four in the Eastern Carpathians (RFE, RCO, RMO, RPO). More distant populations from outside of the Carpathian region were also included as outgroup populations: one from the Bulgarian Rila Mountains (BYU) and one from the Central Estonian Plain (ESE). Our sampling plots are found in natural sites as part of nature reserves, and none of them are affected by forest activity. Accordingly, the Scots pine populations regenerate naturally. Most of the populations persist in ecologically extreme conditions, including dry, sunny outcrops with low soil availability, acidic peatbogs and dry sandy substrates with low nutrient content.
Needles were sampled from mature trees (8–30 from each population) with at least 30-m distance between individuals. However, in some of the populations, samples were taken from shorter inter-individual distances due to relatively small population size (from 0.02 to >5.00 km2) and limited sample availability. Plant material was stored on silica gel and frozen at −80 °C until DNA extraction.
Laboratory methods
Total DNA was extracted from 20 to 25 mg of plant material (1-year-old needles) by using DNeasy Plant Mini Kit (QIAGEN, Valencia, CA, USA). For the first attempt, ten nuclear microsatellites were used (SPAG 7.14, SPAC 11.4, SPAC 11.6 and SPAC 12.5 from Soranzo et al. (1998) and psyl16, psyl17, psyl19, psyl36, psyl42 and psyl57 from Sebastiani et al. (2012)). Later, two inconsistent loci (SPAC 11.6, SPAC 12.5) were excluded from further analysis. The remaining eight markers showed reliable banding patterns and were scored and used for further analysis. These markers proved to be highly polymorphic in our populations. Genotyping of individuals at chloroplast SSR loci was performed with four primer pairs, Pt-30204, Pt-15169, Pt-45002 and Pt-26081 originally developed for Pinus leucodermis (Vendramin et al. 1996). Forward nuclear primers were fluorescently labelled with 6-FAM (SPAG 7.14, SPAC 11.4, psyl17, psyl19, psyl42 and psyl57) and NED (psyl16, psyl36). Chloroplast primers were labelled with HEX (Pt-30204, Pt-26081) and 6-FAM (Pt-15169, Pt-45002). Polymerase chain reaction (PCR) details are provided in the Online Resource. Electrophoresis to detect PCR products were carried out on a 1% (w/v) ethidium bromide-stained agarose gel in 1× TBE buffer. After amplification, strong yields of PCR products were diluted 25 to 30 times for fragment sizing, which was performed on an automated sequencer ABI PRISM 3100 Genetic Analyser (Applied Biosystems, Foster City, CA). Band scoring was analysed using PeakScanner 1.0 (Applied Biosystems 2006), and all size scores were visually checked.
Data analysis
Micro-Checker (Van Oosterhout et al. 2004) was used to test all nSSR loci for null alleles and possible scoring errors derived from large allele dropout and the presence of microsatellite stutter bands. Then, standard population genetic diversity indices (number of alleles (Na), number of effective alleles (Ne), number of private alleles (Np), expected heterozygosity (H e), observed heterozygosity (H o), inbreeding coefficient (F IS)) were calculated for each population using GenAlEx v.6.5 (Peakall and Smouse 2006). For cpSSR loci, haploid genetic diversity (h) and Na, Ne and Np were calculated with GenAlEx v.6.5. For estimates of haplotype diversity indices, haplotype analysis (Eliades and Eliades 2009) was used. Relying on the average squared sum of all allele size differences with the assumption of the stepwise mutation model (Morgante et al. 1998), we calculated mean genetic distance between individuals (D sh 2; Goldstein et al. 1995). In addition, the number of haplotypes per population (A), the number of private haplotypes (P h ), the effective number of haplotypes (N e ) and haplotypic richness (H R ) were estimated using rarefaction (El Mousadik and Petit 1996).
Analysis of molecular variance (AMOVA) implemented in Arlequin v.3.5 software (Excoffier and Lischer 2010) was used for nSSR and cpSSR data to determine the partition of the genetic variation within and among populations. Significance tests were evaluated using a permutation approach with 999 replications. Similarly, Arlequin v.3.5 was used to detect deviation from expectations of Hardy-Weinberg equilibrium (HWE) at nuclear loci. The test of HWE employed a Markov chain approximation (Guo and Thompson 1992). The number of steps after the burn-in period was set to 1,000,000 with a 100,000 dememorization.
To test for correlation between geographical (kilometres) and genetic (Nei’s unbiased genetic distance) distances between pairs of populations at both nSSR and cpSSR markers, dissimilarity matrices were generated and tested for isolation by distance (IBD; Wright 1943) with a Mantel test (Mantel 1967). The analysis was carried out in the online platform of Isolation-by-Distance Web Service (IBDWS) 3.16 (Jensen et al. 2005) and GenAlEx v.6.5, with 9999 and 1000 permutations, respectively.
Different approaches were employed to investigate the spatial genetic structure of the populations. A Bayesian clustering approach implemented in STRUCTURE 2.3.4 (Pritchard et al. 2000) was used to infer groups or subpopulations in the nSSR dataset. We performed the analysis with an admixture model with correlated allele frequencies and a LOCPRIOR setup (according to preliminary test runs). The method uses sampling locations as prior information in the case of a relatively weak signal of structures (Hubisz et al. 2009). K value was set to 1–10 with a burn-in period of 105 steps followed by 106 repetitions of Markov chain Monte Carlo (MCMC). Fifteen repetitions were set for each run. The web-based STRUCTURE HARVESTER (Earl and von Holdt 2012) was used to apply the Evanno method (Evanno et al. 2005) to detect the value of K (the number of genetic groups) that best fit the data. The 15 simulations were averaged using CLUMPP v1.1.2 (Jakobsson and Rosenberg 2007) and represented in the form of bar graphs using POPHELPER (Francis 2016). We used BAPS 6.0 (Corander and Marttinen 2006; Corander et al. 2008a, 2008b) to conduct hierarchical clustering analyses of the chloroplast microsatellite dataset. BAPS was run with the maximal number of groups (K) set to 2–20 (equal or larger than the population number), and each run was replicated five times.
Genetic discontinuities corresponding to the change in genetic variation among populations were identified with BARRIER 2.2 (Manni et al. 2004) on both nuclear and chloroplast SSR datasets. Spatial geographic coordinates were connected with Delaunay triangulation, and the corresponding Voronoi tessellations were projected. To identify the genetic barriers, Monmonier’s maximum difference algorithm (1973) was applied, which traces a barrier along the Voronoi tessellation, starting from the edge for which the distance value is maximal. The boundary proceeds across adjacent edges until the forming boundary reaches the limit of the Delaunay triangulation or closes itself by forming a loop around a population. Within the analysis, barriers were set from 1 to 9. To test the significance of the detected barriers, 1000 resampled bootstrapped (population pairwise) D A genetic distance matrices (Nei’s chord distance; Nei 1978) were calculated in MICROSATELLITE ANALYSER (MSA) software (Dieringer and Schlötterer 2003).
To detect any recent severe reduction in effective population size or possible expansion events in Scots pine populations, BOTTLENECK 1.2.02 was used on the nSSR dataset (Cornuet and Luikart 1996). Bottlenecks cause low-frequency alleles to become transitorily less abundant (<0.1), while more intermediate-frequency alleles increase (Luikart et al. 1998). BOTTLENECK correlates expected heterozygosity (H e) with observed heterozygosity (H o) at mutation-drift equilibrium. The two-phased model (TPM) of mutation was applied as the most appropriate for microsatellite data (Di Rienzo et al. 1994; Piry et al. 1999). We used 5% of multistep changes and a variance among multiple (12) steps (Piry et al. 1999). For each population, 2000 simulations were performed. Significance was assessed using the implemented Wilcoxon sign-rank test, which determines whether or not the average of standardized differences between H o and H e is significantly different from zero (Cornuet and Luikart 1996). Significant heterozygote excess relative to the number of alleles indicates a recent population bottleneck. Additionally, the ‘mode shift’ qualitative descriptor of allele frequency distribution was applied to discriminate bottlenecked populations (Luikart and Cornuet 1998).
Results
Chloroplast microsatellites
All four chloroplast microsatellite markers amplified successfully, and polymorphism was found at all loci (Online Resource Table S1). Haplotype analysis of the SSRs revealed 4 to 13 size variants per locus. A total of 36 size variants at the four loci were identified. These size variants combined into 141 haplotypes, 87 of which were private, having frequencies of <1.0%. The number of haplotypes detected in each population was 3 to 23. Haploid diversity (h) was balanced along the populations (without any outlier value) and ranged from 0.349 in RFE to 0.703 in RBI. Similarly, the number of alleles (Na 2.5–5.0) and the number of effective alleles (Ne 1.706–3.558) were balanced without any outstanding differences. Mean number of private alleles was the highest in SME (1). The overall mean of genetic distance between individuals (D sh 2) was 5.792, but in SME (17.070), RBI (20.893) and RCO (11.986), this value was substantially higher than the average (Online Resource Table S1).
The geographic distribution of the 20 highest frequency haplotypes is reported in Fig. 1. There were differences in haplotype frequencies between the Eastern Carpathian populations on one hand and the Transylvanian Central-Island Mountains (Apuseni) and Southern Carpathian populations on the other. Moreover, Western Carpathian populations showed differences in haplotype proportions and occurrences in haplotypes when the four populations were separated into two groups: High Tatra (SME, SKV) and Lower Tatra (SLI, STU). It is also noticeable that three of the Hungarian populations (HZA, HVE, HFE) harboured only 7–8 of the 20 most common haplotypes. The average occurrence along the range of the 20 common haplotypes was 4–5, with some exceptions.

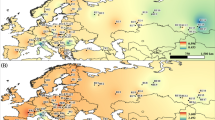
Geographic location and distribution of the cpSSR haplotypes with the frequency of the 20 most common haplotypes among the populations sampled. The slate grey portion of the pie charts represents the remaining, less frequent haplotypes. The acronyms stand for the population code in Table 1. The natural distribution of Scots pine is marked in grey according to the EUFORGEN database, with modifications by the authors. RL Rila Mountains, CE Central Estonian Plain, PB Pannonian Basin, WC Western Carpathians, EC Eastern Carpathians, SC Southern Carpathians and CIM Central-Island Mountains (Apuseni)
Nei’s genetic distance between populations varied from 0.063 to 0.664, with a mean of 0.181. AMOVA analysis including all populations resulted in ΦPT = 0.074 (p < 0.001), meaning that 7% of genetic variance resides among and 93% within populations. Within the Carpathians (excluding ESE and BYU), AMOVA presented ΦPT = 0.081 (p < 0.001). The average gene flow (Nm; calculation based on F ST ) was estimated at 6.272 migrants per generation. The Mantel test revealed significant negative correlation between geographic and genetic distances among populations (r xy −0.176, p < 0.045). The Bayesian approach of population structuring estimated with BAPS identified three main clusters (K = 3; Fig. 2), which formed geographically distinct groups. Two major groups were detected: Populations from western Hungary clustered together with those of the Eastern Carpathians and the Bulgarian population (1), while populations of the Western Carpathians (Tatras) and the Transylvanian Central-Island Mountains (Apuseni) grouped together with the Southern Carpathian and Estonian populations (2). Population SME from the Tatras was separated as a distinct group.

Estimated population structure for K = 3 according to BAPS analysis (Corander and Marttinen 2006; Corander et al. 2008a, 2008b). The acronyms stand for the population code in Table 1. The natural distribution of Scots pine is marked in grey according to the EUFORGEN database, with modifications by the authors. RL Rila Mountains, CE Central Estonian Plain, PB Pannonian Basin, WC Western Carpathians, EC Eastern Carpathians, SC Southern Carpathians and CIM Central-Island Mountains (Apuseni)
The main barriers to gene flow as detected with BARRIER on the cpSSR dataset delimited the Eastern Carpathian populations with highest bootstrap support of 99.5% (Online Resource Fig. S1). Between the Eastern and the Southern Carpathian populations, the barrier was only weak (34.3%). Another strong barrier (with up to 86.1% support) was drawn around the RML population in the Transylvanian Central-Island Mountains (Apuseni). In the High Tatra, SME population also showed genetic discontinuity with a 58% supported barrier.
Nuclear microsatellites
All loci largely conformed to the HWE and showed no significant deviations, although a few populations partially showed minor deviations, which cannot be associated with null alleles or non-neutral behaviour. Micro-Checker test confirmed the lack of null alleles.
A moderate level of intra-population variability was found. The mean of H e was 0.586, which ranged from 0.493 in RMO to 0.648 in HKO. H o was 0.589, ranging from 0.488 in RMO to 0.652 in HKO. The latter HKO population showed the highest value, both in the number of alleles (Na 8.250) and in the number of effective alleles (Ne 4.862). The mean number of private alleles was remarkably high in RPO (1.750), while in the rest of the populations, the overall mean was 0.268. Inbreeding coefficient (F IS) varied greatly, ranging from −0.2411 in RFE to 0.2830 in RPO, with a mean of −0.0329 (Online Resource Table S1).
AMOVA showed high molecular variance within individuals (90%) and relatively low molecular variance among individuals (3%), while among populations, only a 7% variation was observed. Overall, F ST was 0.071 (p < 0.001), while within the Carpathians, F ST was 0.075 (p < 0.001). The number of migrants per generation (Nm) was estimated at 3.247. The Mantel test of IBD yielded no significance value (r xy 0.085, p < 0.233), and the distribution of Nei’s genetic distances over the geographic region did not show limited or restricted gene flow (Online Resource Table S1).
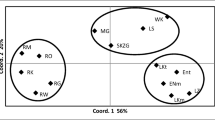
By evaluating nuclear SSR variation with STRUCTURE when K = 2, the two groups were as follows (Fig. 3): (1) Eastern Carpathian populations with the Bulgarian population (BYU) and the northernmost population from Estonia (ESE) and (2) western Hungarian, Western Carpathian populations and the Southern Carpathian population. Populations from the Transylvanian Central-Island Mountains (Apuseni Mts.) were distributed in both clusters: RMH, RML and RBE in cluster 1 and RBI in cluster 2. At K = 3, two peat bog populations (RMO, RPO) from the Eastern Carpathians were differentiated forming cluster 1. Although populations from Bulgaria and Estonia showed grouping with the Eastern Carpathian populations at K = 2 and K = 3, these populations were highly admixed (Fig. 3).

Estimated population structure for K = 2 and K = 3 of assignment analysis performed in STRUCTURE (Pritchard et al. 2000). K = 2 clusters of individuals detected by Evanno et al. (2005) implemented in STRUCTURE HARVESTER (Earl and von Holdt 2012) plotted on a geographic map. The most likely membership to one of the K clusters is presented by the colour of the individual’s thin line. The acronyms stand for the population code in Table 1. The natural distribution of Scots pine is marked in grey according to the EUFORGEN database, with modifications by the authors. RL Rila Mountains, CE Central Estonian Plain, PB Pannonian Basin, WC Western Carpathians, EC Eastern Carpathians, SC Southern Carpathians and CIM Central-Island Mountains (Apuseni)
To identify underlying subclusters, the two clusters (namely cluster 1 and cluster 2) were reanalysed separately (data not shown). Within cluster 1, RFE was separated, while in cluster 2, high admixture was detected without any clear substructuring among the Hungarian, Slovakian and Romanian populations.
BARRIER analysis identified major genetic discontinuities with high bootstrap support (from 70.9 to 100%) around the Eastern Carpathians, separating these populations from the rest (Online Resource Fig. S1). Additionally, the single RCO population from rocky substrate was separated within the Eastern Carpathians with a 99.5% highest support. As in the case of the cpSSR dataset, around the RML population, a strong barrier (up to 92.2% support) was detected around the RML population. All the other barriers between the populations were weak and indicated a non-significant separation with <45% bootstrap support.
The BOTTLENECK analysis showed no evidence of significant excess or deficit of heterozygosity in most populations (19) under the TPM model. As an exception, a recent decline (heterozygote excess) was detected in the RML population from the Central-Island Mountains (Apuseni). This was also supported by the Wilcoxon sign-rank test. Additionally, a shift in the distribution of allele frequencies was detected in the RML population. In the case of non-bottlenecked Scots pine populations, the mode shift indicator test identified a normal L-shaped form of allele frequency distribution as expected in populations that are near to mutation-drift equilibrium. On the basis of the distribution of allele frequencies, we can assume that populations are randomly mating.
Discussion
This study aims to assess the genetic diversity and differentiation, based on nuclear and chloroplast microsatellite marker analysis, of P. sylvestris populations considered to be natural and inhabiting the species distribution range limits along the Carpathians and the Pannonian Basin. Both marker types showed overall high genetic variation along the range studied, suggesting that population fragmentation events might have taken place relatively recently.
Although the two marker types exhibit particular genetic patterns, there were also some congruencies in the detected spatial genetic structures of populations. BAPS and STRUCTURE analysis delimited Western Carpathian populations that proved to be different from those inhabiting the eastern range of the Carpathians (Figs. 2 and 3). Furthermore, Eastern Carpathian populations were differentiated by cpSSR markers from the populations of the Transylvanian Central-Island Mountains (Apuseni) (Figs. 1 and 2). The differentiation in the genetic pattern along the Carpathians has been recognized in earlier studies of other conifer species. In the case of Picea abies or Abies alba, mitochondrial minisatellite regions and nSSRs delimited lineages of the Western and Eastern Carpathians, and accordingly, these patterns suggest different origins of populations from distinct glacial refugia (Tollefsrud et al. 2008; Liepelt et al. 2009; Gömöry et al. 2012). Salix herbacea, an arctic-alpine species from the Carpathian region, showed identical population structure highlighted by BAPS and STRUCTURE and confirmed the distinct origin of the Carpathian populations in question (Alsos et al. 2009). Moreover, our revealed haplotype pattern in Scots pine is highly congruent with that observed by Höhn et al. (2009) for Pinus cembra, where the populations of the Western and the Eastern Carpathians were spatially separated on the basis of chloroplast SSR variation. However, this separation was not significant in the study by Lendvay et al. (2014), which was done using nuclear SSR markers. Based on a cluster analysis of cpSSR haplotype frequencies in P. abies, Bucci and Vendramin (2000) revealed genetic differentiation between Western and South-Eastern Carpathian populations. These findings correspond with the main geobotanical regions described earlier for the Carpathians (Georgescu and Donita 1965; Zemanek 1991; Ronikier 2011).
The Western Carpathian population Medzi bormi (SME) from Slovakia was the most outstanding, showing the overall highest number of private alleles (Np = 1.000) and forming a distinct cluster in the BAPS analysis. Moreover, BARRIER analysis also separated this population from those inhabiting the same range with a moderate bootstrap support (58%). This might be explained by hybrid individuals of Pinus rhaetica (P. sylvestris x Pinus mugo) reported earlier from this peat bog (Staszkiewicz 1994; Kormut’ák et al. 2013). Bottleneck and restricted gene flow was detected by BARRIER in the Mluha population (MLA) from the Transylvanian Central-Island Mountains, which might be a consequence of the decreased population census size where closely related individuals are mating within an isolated stand. On the basis of a very early first description of the Mluha peatbog in which Scots pine is not mentioned (Csató 1885), one plausible explanation would be the recent colonization of the peatbog by this species. Later, a low population size was mentioned by Pacurar et al. (2010). Additionally, the Fantana Brazilor (RFE) population from the Eastern Carpathians appeared as a conspicuously different group in the substructure analysis and also showed a distinct haplotype proportion in the region. It is possible that this stand originates from a distinct refugium or might bear signs of historical human influence.
On the basis of both chloroplast and nuclear microsatellite markers that presented congruent structure with the previously mentioned conifer species, it is likely that the Carpathian populations of Scots pine harbour genetic material originating from at least two separate refugia, dating back to the Pleistocene. One refugium might have been situated around the Eastern Alps and the Hungarian Plain with the Danube region (Cheddadi et al. 2006; Tribsch and Schönswetter 2003), and the other might have existed in the Eastern Carpathians, where a high abundance of fossil pollen remains of diploxyon Pinus species was reported (Feurdean et al. 2011). These two possible refugia were also reported for subalpine and alpine perennial plant species, such as Hipochoeris uniflora (Mráz et al. 2007) and Campanula alpina (Ronikier et al. 2008; Ronikier and Zalewska-Gałosz 2014), which present similar delimitations in population structure and support the North-Eastern Alpine and East Carpathian refugia. Eastern Carpathian populations might have also served as source populations in later Holocene colonization towards northern latitudes as described for other coniferous species (Latałowa and van der Knaap 2006; Feurdean et al. 2007; Tollefsrud et al. 2008). This is probably the case, since the Estonian population and the more southern Bulgarian population clustered together.
Levels of genetic diversity revealed in our study with nSSR markers (mean expected heterozygosity (H e : 0.586) showed similar values as those found in Bulgarian populations (Naydenov et al. 2011), but the values are much lower than those in the more southern peripheral populations from the Apennines and the Southern Alps studied by Scalfi et al. (2009). Likewise, we have found overall higher values compared to those reported by Bernhardsson et al. (2016) from the Romanian-Hungarian region. This might be attributed to the larger sampling area of our study, which included not just the Romanian Carpathians but also the Western Carpathians (the Tatras), and involved a higher number of Hungarian populations. However, it should be noted that this difference can be also the consequence of different marker set, type and number, respectively.
Chloroplast haploid diversity values (h: 0.546) were also lower than the diversity detected by similar markers (Vendramin et al. 1996; Soranzo et al. 1998) at the edge of the range populations in Italy by Scalfi et al. (2009) and in Spain by Robledo-Arnuncio et al. (2005). The lower chloroplast population diversity indices might show signs of isolation and fragmentation of the studied populations as the consequence of restricted gene flow on a regional scale or might be due to the genetic drift, which affects more strongly the populations with decreased census size. As the uniparentaly inherited chloroplast genome is more susceptible to genetic drift compared to the nuclear genome involved in the recombination of the meiosis (Birky et al. 1989; Petit et al. 1993), the low values in cpSSR diversity might show early signs of population fragmentation. Genetic discontinuity was also detected in both datasets with BARRIER analysis.
As expected, a high number of cpSSR haplotypes was detected (36 size variants combined into 141 haplotypes) all over the range studied in the Carpathian populations. A high number of haplotypes was also reported in earlier studies by Naydenov et al. (2005), Robledo-Arnuncio et al. (2005) and Cheddadi et al. (2006), since these microsatellite regions have very high mutation rates (Vendramin and Ziegenhagen 1997; Vendramin et al. 1998; Provan et al. 1998).
In most of the populations studied, we did not find signs of inbreeding, as in our study, F IS values were overall negative (−0.0329), except in the cases of HVE, SME and RPO, for which F IS values were found to be positive (0.184, 0.197 and 0.283). Bernhardsson et al. (2016) reported an overall positive F IS value, potentially as a consequence of artificially maintained and human-restored populations. Alternatively, we consider that in HVE, SME and RPO, the small population distribution area (0.02–4.49 km2) and highly isolated habitat have increased the rate of selfing and might exhibit higher viability of selfed offspring (Savolainen et al. 1992), resulting in a slightly increased positive F IS value. Although small and isolated populations are more vulnerable to inbreeding (Ellstrand and Elam 1993), our overall results regarding most of the population studied are in accordance with earlier statements according to which inbreeding takes generations to develop, and/or even with a restricted gene flow, populations still maintain gene exchange. By estimating gene flow between populations, we detected a relatively high number of possible migrants per generation. Between populations, the value of the number of migrants per generation was fairly large (Nm = 6.272) for chloroplast and moderate (Nm = 3.247) for nSSRs.
In accordance with the lack of signs of inbreeding, BOTTLENECK analysis provided evidence that the Carpathian populations studied are not influenced by a recent genetic bottleneck. Long-lasting signs of bottlenecks require multiple generations to appear. Furthermore, the effects can vary not only based on the reduction size but also depending on the duration period (Busch et al. 2007; Peery et al. 2012). It is most plausible that populations that today are isolated have undergone a recent fragmentation and isolation event. Macrofossil and pollen records indicate that conifer species like P. sylvestris with diploxylon pollen type have survived the LGM in the Carpathians and the Pannonian Basin (Rudner et al. 1995; Rudner and Sümegi 2001; Magyari 2011), and a strong withdrawal and population decline began only between 8000 and 10,000 years BP (or even later, depending on geographic location and elevation) in the Late Glacial/Holocene transition period to mid-Holocene (Tantau et al. 2003, 2006; Feurdean and Bennike 2004; Feurdean et al. 2007, 2012). Transition from coniferous stands to mixed forests has been detected by Mihai et al. (2007), and recently, within the last decades, increasing clear-cutting of the coniferous forests for pasturing has been reported by Motta et al. (2006).
Both marker types in our study presented a relatively high among-population differentiation, as in the cases of other peripheral study sites in the Italian Alps and the Apennines (Scalfi et al. 2009; Belletti et al. 2012). In our results, population differentiation was ΦPT = 0.071 in the case of nuclear and ΦPT = 0.074 for chloroplast SSRs within the range studied. This relatively high differentiation detected might be due to historical demographical events and presumably are not the consequence of pollen exchange restriction and/or effect of geographic isolation caused by the complex architecture of the Carpathians. The barriers identified among regions are not considered impervious, because neither the high level of gene flow nor the low inbreeding values support this. Our estimated degree of genetic differentiation in the Carpathian region might be related to the contact zone that has been established as a consequence of the migration of diverged lineages that survived glaciation in separated refugia and marked the geographical barrier detected.
In conclusion, despite former findings, our microsatellite study of peripheral Scots pine populations from central-eastern Europe have revealed genetic discontinuities along the Carpathian arch. Genetic differentiation of the Eastern Carpathians based on both marker types indicates different origin of these populations and supports the hypothesis of a once existed Pleistocene refugia within the region, also evidenced by some recent palynological records. The genetic structure between the Western and the Eastern Carpathians was revealed by former phylogeographical studies of tree species native to the Carpathians. The fact that the populations in the Western Carpathians are separated from the Eastern Carpathian populations and cluster with the Hungarian populations of the Pannonian Basin indicates another genetic lineage that might have originated from earlier reported refugia of the surroundings of the Eastern Alps and the Hungarian Plain. As natural Scots pine populations from the Carpathians still harbour high gene stocks yet unaffected by isolation and genetic erosion, the populations represent valuable genetic resources of the species. Enhanced by their peripheral position, in addition to the evidence provided by their particular phylogeographic pattern, these stands can also provide insights into the species’ adaptive genetic variation, enriching our understanding of population genetic processes and allowing us to better assess the impact of ongoing climate change.
References
Alsos IG, Alm T, Normand S et al (2009) Past and future range shifts and loss of diversity in dwarf willow (Salix herbacea L.) inferred from genetics, fossils and modelling. Glob Ecol Biogeogr 18:223–239
Belletti P, Ferrazzini D, Piotti A et al (2012) Genetic variation and divergence in Scots pine (Pinus sylvestris L.) within its natural range in Italy. Eur J For Res 131:1127–1138
Bernhardsson C, Floran V, Ganea SL et al (2016) Present genetic structure is congruent with common origin of distant Scots pine populations in its Romanian distribution. Forest Ecol Manag 361:131–141
Birks HH, Ammann B (2000) Two terrestrial records of rapid climatic change during the glacial–Holocene transition (14,000–9,000 calendar years BP) from Europe. Proc Natl Acad Sci U S A 97:1390–1394
Birky CW Jr, Fuerst P, Maruyama T (1989) Organelle gene diversity under migration, mutation, and drift: equilibrium expectations, approach to equilibrium, effects of heteroplasmic cells, and comparison to nuclear genes. Genetics 121:613–627
Bradshaw RH (2004) Past anthropogenic influence on European forests and some possible genetic consequences. Forest Ecol Manag 197:203–212
Bucci G, Vendramin GG (2000) Delineation of genetic zones in the European Norway spruce natural range: preliminary evidence. Mol Ecol 9:923–934
Busch JD, Waser PM, DeWoody JA (2007) Recent demographic bottlenecks are not accompanied by a genetic signature in banner-tailed kangaroo rats (Dipodomys spectabilis). Mol Ecol 16:2450–2462
Čelepirović N, Ivanković M, Gradečki-Poštenjak M et al (2009) Review of investigation of variability of nad1 gene intron B/C of mitochondrial genome in Scots pine (Pinus sylvestris L.) Period Biol 111:453–457
Chapin FS III, Zavaleta ES, Eviner VT et al (2000) Consequences of changing biodiversity. Nature 405:234–242
Cheddadi R, Vendramin GG, Litt T et al (2006) Imprints of glacial refugia in the modern genetic diversity of Pinus sylvestris. Glob Ecol Biogeogr 15:271–282
Chhatre VE, Rajora OP (2014) Genetic divergence and signatures of natural selection in marginal populations of a keystone, long-lived conifer, eastern white pine (Pinus strobus) from northern Ontario. PLoS One 9:e97291
Corander J, Marttinen P (2006) Bayesian identification of admixture events using multi-locus molecular markers. Mol Ecol 15:2833–2843
Corander J, Marttinen P, Sirén J et al (2008a) Enhanced Bayesian modelling in BAPS software for learning genetic structures of populations. BMC Bioinformatics 9:539
Corander J, Sirén J, Arjas E (2008b) Bayesian spatial modelling of genetic population structure. Comput Stat 23:111–129
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Crespi BJ (2000) The evolution of maladaptation. Heredity 84:623–629
Csató J (1885) A Mluha nevű tó (Teu Mluhi) és viránya. [in English: plants of lake Mluha (Teu Mluhi)]. Magyar Botanikai Lapok 9:1–8
Di Rienzo A, Peterson AC, Garza JC et al (1994) Mutational processes of simple sequence repeat loci in human populations. Proc Natl Acad Sci U S A 91:3166–3170
Dieringer D, Schlötterer C (2003) Microsatellite analyser (MSA): a platform independent analysis tool for large microsatellite data sets. Mol Ecol Notes 3:167–169
Earl DA, von Holdt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
El Mousadik A, Petit RJ (1996) High level of genetic differentiation for allelic richness among populations of the argan tree [Argania spinosa (L.) Skeels] endemic to Morocco. Theor Appl Genet 92:832–839
Eliades NG, Eliades DG (2009) Haplotype Analysis: software for analysis of haplotype data. Forest Göttingen (Germany): Genetics and Forest Tree Breeding, Georg-August University Göttingen. Distributed by the authors. http://www.uni-goettingen.de/en/134935.html. Accessed: 05 October 2016
Ellstrand NC, Elam DR (1993) Population genetic consequences of small population size: implications for plant conservation. Annu Rev Ecol Syst 24:217–242
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Ewers RM, Didham RK (2006) Confounding factors in the detection of species responses to habitat fragmentation. Biol Rev 81:117–142
Excoffier L, Lischer HE (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Fahrig L (2003) Effects of habitat fragmentation on biodiversity. Annu Rev Ecol Evol Syst 34:487–515
Fekete L, Blattny T (1913) Az erdészeti jelentőségú fák és cserjék elterjedése a Magyar Állam területén (The distribution of forest trees and shrubs in Hungary). Erdészeti Lapok 52:743–758
Fekete G, Molnár Z, Magyari E et al (2014) A new framework for understanding Pannonian vegetation patterns: regularities, deviations and uniqueness. Community Ecol 15:12–26
Feurdean A, Bennike O (2004) Late Quaternary palaeoecological and palaeoclimatological reconstruction in the Gutaiului Mountains, northwest Romania. J Quaternary Sci 19:809–827
Feurdean A, Wohlfarth B, Björkman L et al (2007) The influence of refugial population on Lateglacial and early Holocene vegetational changes in Romania. Rev Palaeobot Palyno 145:305–320
Feurdean A, Tanţău I, Fărcaş S (2011) Holocene variability in the range distribution and abundance of Pinus, Picea abies, and Quercus in Romania; implications for their current status. Quaternary Sci Rev 30:3060–3075
Feurdean A, Björkman L, Wohlfarth B (2012) A paleoecological reconstruction of the Late Glacial and Holocene based on multidisciplinary studies at Steregoiu site (Gutai Mts., Romania). Studia UBB Geologia 46:125–140
Francis RM (2016) POPHELPER: an R package and web app to analyse and visualise population structure. Mol Ecol Res. doi:10.1111/1755-0998.12509
Georgescu GG, Donita L (1965) La division floristique de Roumanie. I Rev Rou Biol, Ser Bot 10:357–369
Giertych M, Mátyás C (2013) Genetics of Scots pine. Amsterdam, Elsevier 3:19–76
Goldstein DB, Linares AR, Cavalli-Sforza LL et al (1995) An evaluation of genetic distances for use with microsatellite loci. Genetics 139:463–471
Gömöry D, Paule L, Krajmerová D et al (2012) Admixture of genetic lineages of different glacial origin: a case study of Abies alba Mill. in the Carpathians. Plant Syst Evol 298:703–712
Guo SW, Thompson EA (1992) Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics 48:361–372
Höhn M, Gugerli F, Abran P et al (2009) Variation in the chloroplast DNA of Swiss stone pine (Pinus cembra L.) reflects contrasting post-glacial history of populations from the Carpathians and the Alps. J Biogeogr 36:1798–1806
Hubisz MA, Falush D, Stephens M et al (2009) Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour 9:1322–1332
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806
Jankovská V, Pokorný P (2008) Forest vegetation of the las full-glacial period in the Western Carpathians (Slovakia and Czech Republic). Preslia 80:307–324
Jensen JL, Bohonak AJ, Kelley ST (2005) Isolation by distance, web service. BMC Genet 6:13
Kieliszewska-Rokicka B (1981) Geographic and climatic differentiation of electrophoretic forms of esterase, glutamate dehydrogenase and peroxidase in Scots pine tissues. Acta Soc Bot Pol 50:481–492
Kormut’ák A, Camek V, Branná M et al (2013) Introgressive hybridization between Scots pine and mountain dwarf pine at two localities of northern Slovakia. Folia Oecol 40:201
Kramer AT, Ison JL, Ashley MV, Howe HF (2008) The paradox of forest fragmentation genetics. Conserv Biol 22:878–885
Kremer A, Ronce O, Robledo-Arnuncio JJ et al (2012) Long-distance gene flow and adaptation of forest trees to rapid climate change. Ecol Lett 15:378–392
Labra M, Grassi F, Sgorbati S et al (2006) Distribution of genetic variability in southern populations of Scots pine (Pinus sylvestris L.) from the Alps to the Apennines. Flora 201:468–476
Latałowa M, van der Knaap WO (2006) Late Quaternary expansion of Norway spruce Picea abies (L.) Karst. in Europe according to pollen data. Quaternary Sci Rev 25:2780–2805
Lendvay M, Höhn M, Brodbeck S, Mîndrescu M, Gugerli F (2014) Genetic structure in Pinus cembra from the Carpathian Mountains inferred from nuclear and chloroplast microsatellites confirms post-glacial range contraction and identifies introduced individuals. Tree Genetics and Genome 10(5):1419–1433
Lesica P, Allendorf FW (1995) When are peripheral populations valuable for conservation? Conserv Biol 9:753–760
Liepelt S, Cheddadi R, de Beaulieu JL et al (2009) Postglacial range expansion and its genetic imprints in Abies alba (Mill.)—a synthesis from palaeobotanic and genetic data. Rev Palaeobot Palyno 153:139–149
Lönn M, Prentice HC (2002) Gene diversity and demographic turnover in central and peripheral populations of the perennial herb Gypsophila fastigiata. Oikos 99:489–498
Luikart G, Cornuet JM (1998) Empirical evaluation of a test for identifying recently bottlenecked populations from allele frequency data. Conserv Biol 12:228–237
Luikart G, Allendorf FW, Cornuet JM et al (1998) Distortion of allele frequency distributions provided a test for recent population bottlenecks. J Hered 89:238–247
Magyari EK (2011) Late Quaternary vegetation history in the Hortobágy steppe and Middle Tisza floodplain, NE Hungary. Studia Bot Hung 42:185–203
Magyari EK, Kuneš P, Jakab G et al (2014) Late Pleniglacial vegetation in eastern-central Europe: are there modern analogues in Siberia? Quaternary Sci Rev 95:60–79
Manni F, Guerard E, Heyer E (2004) Geographic patterns of (genetic, morphologic, linguistic) variation: how barriers can be detected by using Monmonier’s algorithm. Human Biol 76:173–190
Mantel N (1967) The detection of disease clustering and a generalized regression approach. Cancer Res 27:209–220
Matías L, Jump AS (2012) Interactions between growth, demography and biotic interactions in determining species range limits in a warming world: the case of Pinus sylvestris. Forest Ecol Manag 282:10–22
Mejnartowicz L (1979) Genetic variation in some isoenzyme loci in Scots pine (Pinus sylvestris L.) populations. Arboretum Kornickie 24:1–1
Mihai B, Savulescu I, Sandric I (2007) Change detection analysis (1986-2002) of vegetation cover in Romania. Mt Res Dev 27:250–258
Morgante M, Felice N, Vendramin GG (1998) Analysis of hypervariable chloroplast microsatellites in Pinus halepensis reveals a dramatic genetic bottleneck. In: Karp A, Isaac PG, Ingram DS (eds) Molecular tools for screening biodiversity. Springer, Netherlands, pp 407–412
Motta R, Morales M, Nola P (2006) Human land-use, forest dynamics and tree growth at the treeline in the Western Italian Alps. Ann For Sci 63:739–747
Mráz P, Gaudeul M, Rioux D et al (2007) Genetic structure of Hypochaeris uniflora (Asteraceae) suggests vicariance in the Carpathians and rapid post-glacial colonization of the Alps from an eastern Alpine refugium. J Biogeogr 34:2100–2114
Müller-Starck G, Baradat P, Bergmann F (1992) Genetic variation within European tree species. New Forest 6:23–47
Naydenov KD, Tremblay FM, Alexandrov A et al (2005) Structure of Pinus sylvestris L. populations in Bulgaria revealed by chloroplast microsatellites and terpenes analysis: provenance tests. Biochem Syst Ecol 33:1226–1245
Naydenov KD, Naydenov MK, Tremblay F et al (2011) Patterns of genetic diversity that result from bottlenecks in Scots pine and the implications for local genetic conservation and management practices in Bulgaria. New Forest 42:179–193
Nei M (1978) Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89:583–590
Pacurar I, Clapa D, Sotropa A (2010) Research on Mluha peatland. Apuseni Mts Agricultura Stiinta si Practica 3-4:75–76
Peakall ROD, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Peery MZ, Kirby R, Reid BN et al (2012) Reliability of genetic bottleneck tests for detecting recent population declines. Mol Ecol 21:3403–3418
Petit RJ, Hampe A (2006) Some evolutionary consequences of being a tree. Annu Rev Ecol Evol Syst 37:187–214
Petit RJ, Kremer A, Wagner DB (1993) Finite island model for organelle and nuclear genes in plants. Heredity 71:630–641
Piry S, Luikart G, Cornuet JM (1999) BOTTLENECK: a computer program for detecting recent reductions in the effective population size using allele frequency data. J Hered 90:502–503
Pócs T (1960) Die zonalen Waldgesellschaften Südwestungarns. [in English: the zonal forest communities of southwestern Hungary]. Acta Bot Hung 6:75–105
Pravdin LF (1969) Scots pine: variation, intraspecific taxonomy and selection. Annarbor Humphrey Science Publishers Ltd., London
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Provan J, Soranzo N, Wilson NJ et al (1998) Gene–pool variation in Caledonian and European Scots pine (Pinus sylvestris L.) revealed by chloroplast simple–sequence repeats. Proc R Soc Lond B Bio 265:1697–1705
Robledo-Arnuncio JJ, Collada C, Alia R et al (2005) Genetic structure of montane isolates of Pinus sylvestris L. in a Mediterranean refugial area. J Biogeogr 32:595–605
Ronikier M (2011) Biogeography of high-mountain plants in the Carpathians: an emerging phylogeographical perspective. Taxon 60:373–389
Ronikier M, Zalewska-Gałosz J (2014) Independent evolutionary history between the Balkan ranges and more northerly mountains in Campanula alpina s.l. (Campanulaceae): genetic divergence and morphological segregation of taxa. Taxon 63:116–131
Ronikier M, Cieślak E, Korbecka G (2008) High genetic differentiation in the alpine plant Campanula alpina Jacq. (Campanulaceae): evidence for glacial survival in several Carpathian regions and long-term isolation between the Carpathians and the Alps. Mol Ecol 17:1763–1775
Rudner E, Sümegi P (2001) Recurring taiga forest-steppe habitats in the Carpathian Basin in the Upper Weichselian. Quatern Int 76:177–189
Rudner E, Sümegi P, Tóth I et al (1995) The vegetation of the upper-Weichselian in the central and southern part of the Great Hungarian Plain. Book of abstracts, 7th European Ecological Congress, pp 108.
Rull V (2009) Microrefugia. J Biogeogr 36:481–484
Rull V (2010) On microrefugia and cryptic refugia. J Biogeogr 37:1623–1625
Savolainen O, Pyhäjärvi T (2007) Genomic diversity in forest trees. Curr Opin Plant Biol 10:162–167
Savolainen O, Karkkainen K, Kuittinen H (1992) Estimating numbers of embryonic lethals in conifers. Heredity 69:308–314
Scalfi M, Piotti A, Rossi M et al (2009) Genetic variability of Italian southern Scots pine (Pinus sylvestris L.) populations: the rear edge of the range. Eur J For Res 128:377–386
Sebastiani F, Pinzauti F, Kujala ST et al (2012) Novel polymorphic nuclear microsatellite markers for Pinus sylvestris L. Conserv Genet Resour 4:231–234
Sommer RS, Zachos FE (2009) Fossil evidence and phylogeography of temperate species:‘glacial refugia’ and post-glacial recolonization. J Biogeogr 36:2013–2020
Soranzo N, Provan J, Powell W (1998) Characterization of microsatellite loci in Pinus sylvestris L. Mol Ecol 7:1260–1261
Staszkiewicz J (1994) Differentiation of population of Pinus x rhaetica (Pinaceae) from the “Medzi bormi” Nature Reserve of Slovakia. Frag Floris Geobot Pol 1:223–233
Sykes MT, Prentice IC, Cramer W (1996) A bioclimatic model for the potential distributions of north European tree species under present and future climates. J Biogeogr 23:203–233
Tantau I, Reille M, de Beaulieu JL et al (2003) Vegetation history in the Eastern Romanian Carpathians: pollen analysis of two sequences from the Mohoş crater. Veg Hist Archaeobot 12:113–125
Tantau I, Reille M, de Beaulieu JL et al (2006) Late Glacial and Holocene vegetation history in the southern part of Transylvania (Romania): pollen analysis of two sequences from Avrig. J Quaternary Sci 21:49–62
Tollefsrud MM, Kissling R, Gugerli F et al (2008) Genetic consequences of glacial survival and postglacial colonization in Norway spruce: combined analysis of mitochondrial DNA and fossil pollen. Mol Ecol 17:4134–4150
Tribsch A, Schönswetter P (2003) Patterns of endemism and comparative phylogeography confirm palaeoenvironmental evidence for Pleistocene refugia in the Eastern Alps. Taxon 52:477–497
Van Oosterhout C, Hutchinson WF, Wills DP et al (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Vendramin GG, Ziegenhagen B (1997) Characterisation and inheritance of polymorphic plastid microsatellites in Abies. Genome 40:857–864
Vendramin GG, Lelli L, Rossi P et al (1996) A set of primers for the amplification of 20 chloroplast microsatellites in Pinaceae. Mol Ecol 5:595–598
Vendramin GG, Anzidei M, Madaghiele A et al (1998) Distribution of genetic diversity in Pinus pinaster Ait. as revealed by chloroplast microsatellites. Theor Appl Genet 97:456–463
Wachowiak W, Salmela MJ, Ennos RA et al (2011) High genetic diversity at the extreme range edge: nucleotide variation at nuclear loci in Scots pine (Pinus sylvestris L.) in Scotland. Heredity 106:775–787
Willis KJ (1994) The vegetational history of the Balkans. Quaternary Sci Rev 13:769–788
Willis KJ, Sümegi P, Braun M et al (1995) The late Quaternary environmental history of Bátorliget, NE Hungary. Palaeogeogr Palaeoclimatol Palaeoecol 118:25–47
Willis KJ, Bennett KD, Birks HJB (1998) The late Quaternary dynamics of pines in Europe. In: Richardson DM (ed) Ecology and Biogeography of Pinus. Cambridge University Press, Cambridge, pp 107–121
Wright S (1943) Isolation by distance. Genetics 28:114
Zemanek B (1991) The phytogeographical boundary between the East and West Carpathians-past and present. Thaiszia 1:59–67
Acknowledgements
The authors are grateful to Tamás Pócs (Hungary), Ivan Iliev (Bulgaria) and Tibor Baranec (Slovakia) for the assistance they offered with the collection of plant material and for the valuable discussions in the course of which they shared their insights. We also wish to thank the Associate Editor for helpful comments and the Anonymous Reviewers who helped us improve the manuscript. We also thank EUFORGEN database for providing the distribution map of Scots pine. This work was supported by National Research, Development and Innovation Office, Hungary, by a grant of the Hungarian Scientific Research Fund [OTKA K101600].
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by F. Gugerli
Electronic supplementary material
ESM 1
(DOCX 484 kb).
Data archiving statement
We are in the process of archiving our microsatellite genotype data in an online database. We will provide details as soon as the process has been completed. Other additional data may be requested from the corresponding author (toth.endre@kertk.szie.hu).
Rights and permissions
About this article

Cite this article
Tóth, E.G., Vendramin, G.G., Bagnoli, F. et al. High genetic diversity and distinct origin of recently fragmented Scots pine (Pinus sylvestris L.) populations along the Carpathians and the Pannonian Basin. Tree Genetics & Genomes 13, 47 (2017). https://doi.org/10.1007/s11295-017-1137-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-017-1137-9