
Abstract
Understanding geographical pattern of genetic diversity and population structure is of great importance for formulating conservation and utilization strategies. In this study, we investigated the genetic diversity and population structure of 28 natural populations of Castanea mollissima in China using eight nuclear and six chloroplast microsatellite makers (nSSRs and cpSSRs). Populations from central China harbored the highest genetic diversity at both nSSR and cpSSR markers (nSSR: H E = 0.705; cpSSR: H = 0.461). The standardized measure of genetic differentiation estimated as G′ ST was 0.447 for nSSR and 0.803 for cpSSR, respectively. The G′ST-based pollen to seed flow ratio is 3.043, indicating that pollen flow is not extensive among C. mollissima populations. No obvious population genetic structure by geographical locations was found by STRUCTURE analysis based on nSSR data, and similarly, no signal of phylogeographic structure was detected for cpSSR analysis. Five boundaries defining zones of maximum genetic differences within the network of the C. mollissima populations were found, and the locations of those barriers were consistent with those of four mountains, i.e., Daloushan Mountain, Dabashan Mountain, Wushan Mountain, and Qingliangfeng Mountain, indicating that those mountains might act as genetic barriers obstructing the genetic exchange among natural C. mollissima populations. These results provide valuable baseline data for conservation and utilization of this species.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chinese chestnut (Castanea mollissima) is naturally distributed in a large area across temperate to sub-tropical regions in China. In central China, C. mollissima can still be found as a key species in natural forests, although it has been utilized and cultivated for nuts and timber for at least 2,000 years. Together with European chestnut (Castanea sativa) and Japanese chestnut (Castanea crenata), Chinese chestnut is a major cultivated species for nut production in the world. However, the production of European chestnuts decreased dramatically since 20th century due to the severe strike of chestnut blight (Cryphonectria parasitica) and ink disease (Phytophthora cinnamoni). Similarly, American chestnut (Castanea dentata), once served as a keystone species in forest ecosystem in Eastern North America, was virtually driven to extinction since the breakout of chestnut blight in the first half of 20th century (Burnham 1988). Chinese chestnut has been proven as important genetic resource for resistance to chestnut blight and ink disease (Anagnostakis 1992) and was extensively used in many breeding programs for disease resistance (Kubisiak et al. 1997; Barakat et al. 2009; Wheeler and Sederoff 2009). Chinese chestnut also plays a critical role for the genetic improvement of cultivated chestnuts in the world (Huang 1998).
Despite the multipurpose characteristics of the species, natural genetic variation of chestnut in China is not fully understood. Genetic diversity is crucial for a species to respond and adapt to environmental changes (Jump and Penuelas 2005). The evolution of disease resistance depends strongly both on the amount of genetic variation on which selection can engage (Crow 2002) and on the genetic structure which play an important role in the evolutionary trajectory of the host response (Parker and Gilbert 2004). Therefore, understanding the variability and genetic structure of natural chestnut populations is of fundamental importance to field biologists with interests in demographic processes that have shaped plant populations, to conservation managers who need indications of conservation priorities in this species, and to pathologists who seek natural variation in resistance to diseases.
Although population genetics of C. sativa and C. crenata have been extensively investigated using molecular markers such as allozyme, RAPD, AFLP, and microsatellite (Villani et al. 1991; Huang et al. 1994; Huang et al. 1998; Fineschi et al. 2000; Dane et al. 2003; Tanaka et al. 2005; Mattioni et al. 2008; Martin et al. 2010; Martín et al. 2012), little is known about natural genetic diversity of Chinese chestnut, except for allozyme diversity (Lang and Huang 1999). However, due to the limited variation displayed by the allozyme markers used, the obtained information was incomplete. Recently, the combination of nuclear and chloroplast microsatellite markers has been increasingly used in plant population genetic studies (Newton et al. 2002; Fontaine et al. 2004; Takahashi et al. 2008; Martins et al. 2011). The joint use of molecular markers derived from different genomes provides a more comprehensive approach to study population structure and insights into demographic history and dynamics, particularly for comparisons of maternally inherited organelle and biparentally inherited nuclear markers (Petit et al. 2005).
In the present study, we used nuclear and chloroplast microsatellite markers to investigate a full range wide genetic diversity and structure of natural C. mollissima populations with specific aims to (1) infer the demographical history of the species, (2) estimate levels of genetic variation and population differentiation, (3) identify spatial population structure and dispersal barriers across the populations.
Material and methods
Sample collection and DNA extraction
An exhaustive survey and sampling for the whole range of the natural distribution of C. mollissima was conducted in 2007–2008, and 28 natural populations were sampled from Eastern, Central, Southwestern and Northwestern China (Fig. 1). The sampling scheme covered the full natural range of this species. Details of population codes, locations, and sample sizes were summarized in Table 1. Fresh leaves were collected and dried in silica gel, or 1-year-old twigs were sampled and stored at 4 °C. The twigs were soaked in water at room temperature for leafing next year. Total genomic DNA was extracted from leaf tissue using a modified 2× CTAB protocol (Doyle and Doyle 1987).

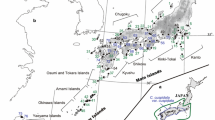
Map showing the locations of sampled populations and geographic distributions of the chloroplast haplotypes found in Castanea mollissima. Populations correspond to those detailed in Table 1
Molecular markers procedure
We selected eight polymorphic nuclear microsatellite markers (nSSR) based on previous experiments, including CsCAT3, CsCAT5, CsCAT8, CsCAT14, CsCAT41 from C. sativa (Marinoni et al. 2003) and KT001b, KT008a, KT020a from C. crenata (Yamamoto et al. 2003). These eight nuclear microsatellite markers are not linked and conform to Hardy–Weinberg equilibrium as proved by previous studies (Wang et al. 2008). For chloroplast microsatellite markers (cpSSR), seven polymorphic markers were chosen after a initial screening polymorphism, including Cmcs2, Cmcs7, Cmcs8, Cmcs12, and Cmcs14 developed for Fagacea (Sebastiani et al. 2004) and udt3 and udt5 identified in Quercus (Deguilloux et al. 2003). PCR amplification was performed in a final volume of 10 μl following procedures described in original papers with minor modifications. Amplified products were separated on a 6 % denaturing polyacrylamide gel using silver staining. Silver staining was conducted according to Sanguinetti et al. (1994). A 25-bp DNA ladder (Promega) was used to identify alleles. To investigate allele-size variation at the seven chloroplast microsatellite loci, one sample of PCR products for each allele at each locus was subjected to DNA sequencing on an ABI 3730xl DNA Analyzer.
Data analysis
Nuclear microsatellite data
The following parameters for revealing genetic diversity in each population were estimated: mean number of alleles per locus (A), mean effective number of alleles per locus (A E), observed heterozygosity (H O), and expected heterozygosity (H E) using GenALEx 6 (Peakall and Smouse 2006). Allelic richness (A R) (El Mousadik and Petit 1996) and within-population fixation index (F IS) were calculated, and the deviation of the fixation indices from Hardy–Weinberg equilibrium (HWE) was tested using FSTAT 2.9.3 (Goudet 2001). The frequency of null alleles was estimated using FreeNA (Chapuis and Estoup 2007).
The global and pairwise population differentiation was evaluated by F ST (infinite allele model, IAM) and R ST (stepwise mutation model, SMM) using FSTAT 2.9.3 and RSTCALC (Goodman 1997), respectively. Statistical significance of the estimates was determined with 1,000 randomizations. Comparison of F ST–R ST was tested based on 1,000 random permutations using SPAGeDi 1.3a (Hardy and Vekemans 2002). Since absolute values of the population differentiation parameters F ST or its relatives depend on the level of genetic variation (Hedrick 2005), we also calculated the standardized measure of genetic differentiation G′ ST (Hedrick 2005) using SMOGD 1.2.5 (Crawford 2010).
BOTTLENECK 1.2.02 (Piry et al. 1999) was used to test deviation from mutation-drift equilibrium, which is expected if populations have experienced a recent demographic decline. To test statistical significance, we used the Wilcoxon signed-rank test under the stepwise mutation model (SMM) and the infinite allele model (IAM). For each mutational model, 1,000 replicates were performed. Due to the small sample sizes of some populations, this test was only performed on populations with sample size N ≥ 16.
Bayesian clustering approach implemented in STRUCTURE 2.3.3 (Pritchard et al. 2000) was used to detect population structure and estimate the number of populations (K) and to assign individuals to one or more of these populations (K). These analyses were based on an admixture ancestry model with correlated allele frequencies and no prior information for population origin. The number of genetically distinct clusters (K) was set to vary from 1 to 10. The model was run ten independent simulations for each K using a burn-in length of 10,000 and a run length of 100,000 iterations. The ∆K statistics, based on the rate of change of log likelihood of data [L (K)] between successive K values was used to select the optimal K followed Evanno et al. (2005).
Grouping populations that were geographically homogeneous and maximally differentiated from each other was also performed using spatial analysis of molecular variance (SAMOVA) (Dupanloup et al. 2002). SAMOVA is based on a simulated annealing procedure that designated to maximize the F CT (an indicator of genetic differentiation among population groups) and the proportion of total genetic variance due to differences between groups of populations. The algorithm was performed by software SAMOVA 1.0 (Dupanloup et al. 2002), which identified the number (K) of groups by the largest F CT value. The value of K was user-defined and set between two and eight, with 100 independent simulated annealing processes in each run.
Genetic distances between each population were estimated by F ST/(1–F ST) using SPAGeDi 1.3a. Latitude and longitude coordinates for each population were used to calculated pairwise Euclidean geographical distances between each population. Mantel test (Mantel 1967) with 10,000 matrix randomization was used to assess the significance of correlations between genetic distances and natural logarithm values of geographical distances using the program IBD 1.52 (Bohonak 2002).
Chloroplast microsatellite data
Length variants at each locus were combined into haplotypes. The following population diversity indices were calculated for each population: number of haplotypes (N), effective number of haplotypes (Ne = 1/∑p i 2), number of private haplotypes (Np), and Nei's haplotype diversity (H = [n/(n − 1)][1 − ∑p i 2]), where for both equations, n is the number of individuals analyzed and p i is the frequency of the ith haplotype in a population (Nei 1987). Allelic richness (A R) was calculated by Contrib 1.02 (Petit et al. 1998). Furthermore, mean within-population genetic diversity, species total genetic diversity were calculated treating alleles as unordered (h S and h T) and ordered (v S and v T), following the method of Pons and Petit (1996) using Permut (www.pierroton.inra.fr/genetics/labo/Software/PermutCpSSR).
In order to investigate the relationships between haplotypes, a phylogenetic network tree was constructed using the median-joining model implemented in NETWORK 4.6 (www.fluxus-engineering.com). The presence of a phylogeographic structure was tested by comparing R ST to pRST (permuted) after 1,000 random permutations using the SPAGeDi 1.3a.
SAMOVA analysis was performed as described above. Additionally, the genetic boundaries were identified with the software BARRIER 2.2 (Manni et al. 2004) using Monmonier's algorithm (Monmonier 1973) based on unbootstrapped single matrix and 100 bootstrap matrices of DA standard genetic distance (Nei et al. 1983). DA standard genetic distance was generated using MICROSATELLITE ANALYZER (MSA) software (Dieringer and Schlötterer 2003). Genetic differentiation and IBD test were analyzed with the same procedure as described in nSSR data.
Results
Nuclear genetic diversity and population bottleneck tests
The eight microsatellite markers used in this study generated a total of 128 alleles across the 849 individuals of C. mollissima. The number of alleles per locus ranged from 10 at locus CsCAT41 to 22 at locus CsCAT5, with a mean number of 16 per locus. Indices of genetic diversity for each population and each region were summarized in Table 2. Mean number of alleles per locus (A) and mean effective number of alleles per locus (A E) for each population ranged from 5.0 to 10.4 and 2.9 to 5.1, respectively. Allelic richness (A R) for each population ranged from 4.6 to 6.6. Mean observed heterozygosity (H O) for each population ranged from 0.497 to 0.688 and mean expected heterozygosity (H E) from 0.600 to 0.759. Populations from Central China exhibited the highest level of genetic diversity (A = 8.1, A E = 4.3, A R = 5.8, A R = 5.8, H O = 0.618), while populations from Southwestern China showed the lowest level of genetic diversity (A = 6.6, A E = 3.4, A R = 4.9, H O = 0.559, H E = 0.640), and moderate level were found in populations from Eastern and Northwestern China. The F IS values were consistently positive due to slight heterozygote deficits found in all populations. Of the 224 locus–population combinations, 41 showed significant deviation from Hardy–Weinberg equilibrium (HWE) at the 5 % level (data not shown), while 18 populations were significantly deviated from HWE at P < 0.05 when loci were combined after applying a Bonferroni correction for multiple tests (Table 2). The average frequencies of null alleles per locus range from 0.025 for locus CsCAT41 to 0.106 for locus CsCAT5.
Theoretically, populations that have undergone recent decline of population size will exhibit deviations from mutation-drift equilibrium and exhibit transient excess of expected heterozygosity (Luikart and Cornuet 1998). No population displayed significant deviation from mutation-drift equilibrium under the SMM; however, eight populations deviated significantly under the IAM (Table 1).
Haplotype variations and relationships
The results of sequencing indicated that variations in allele size were due to the increase or decrease in the number of repeats (Cmcs2, Cmcs7, udt3, udt5), except in some cases where differences were also related to indels in the flanking regions (Cmcs8, Cmcs12). However, for locus Cmcs14, the only two alleles were characterized by a difference of consecutive 41-nucleotide deletion leading to the lack of microsatellite in the shorter allele. Therefore, the Cmcs14 locus was excluded from further analysis. The remaining six chloroplast microsatellite loci assayed for 659 C. mollissima individuals produced a total of 17 different alleles. Because the chloroplast genome does not recombine, a unique combination of size variants (alleles) across the microsatellite regions (loci) was defined as a different haplotype. The combination of 17 different alleles produced 39 unique haplotypes. The haplotype frequency varied greatly, with nine haplotypes being individual-private and 24 haplotypes being population-private. The number of haplotypes per population varied between one and eight. The most abundant haplotype was H35 which was found in 175 individuals from 13 populations (overall frequency of 26.56 % averaged over the total samples). Haplotypes diversity varied greatly across the populations (Table 2). Populations from Central China exhibited the highest level of haplotype diversity (N = 3.9, Ne = 2.1, A R = 1.7, H = 0.461), among which the GM population showed the highest haplotype diversity (N = 8, Ne = 3.6, A R = 3.4, H = 0.749). Populations from Northwestern China showed the lowest haplotype diversity (Ne = 1.6, Np = 0.3, A R = 1.2, H = 0.359). Overall, there was low within-population diversity (h S = 0.433, v S = 0.278), but high total diversity (h T = 0.895, v T = 0.902). The median-joining network produced a complex pattern and no obvious phylogeographic structure could be found by assigning geographic locations to the haplotypes (Fig. 2). The distribution of haplotypes across the range of C. mollissima also showed no obvious geographic structure (Fig. 1). This was further confirmed as the pRST values after 1,000 random permutations was significantly higher than R ST values (pRST = 0.718 > R ST = 0.627, P = 0.014), suggesting a lack of signal of phylogeographic structure.

Median-joining haplotype network for Castanea mollissima populations based on chloroplast microsatellites. Circles of the same color represent haplotypes from the same geographical region. The size of the circle is proportional to the frequency of the haplotype. Open circles indicate unobserved haplotypes. Number on the branch represent more than one mutation step
Population genetic differentiations
The global genetic differentiation across all the populations of C. mollissima estimated as F ST and R ST were 0.120 and 0.194 for nSSR and 0.678 and 0.629 for cpSSR, respectively. For nSSR, the R ST value was significantly higher than F ST after 1000 random permutations (R ST = 0.194 > pRST = 0.108, P = 0.000). Most pairwise values of F ST for nSSR (0.016–0.278) and cpSSR (0.011–0.967) as well as the pairwise R ST for nSSR (0.001–0.563) and cpSSR (0.003–0.966) were significant (P < 0.05) (data not shown). Standardized measure of genetic differentiation (G′ ST) averaged over loci were 0.447 for nSSR and 0.803 for cpSSR, respectively. The pollen to seed migration ratio estimated according to Ennos (1994), i.e., (pollen flow)/(seed flow) = {[(1/ G′ STb) − 1] − 2[(1/ G′ STm) − 1]}/[(1/G′ STm) − 1] is 3.043.
Population structure revealed by nSSRs and cpSSRs
To investigate population structure, a Bayesian population structure analysis was first performed including all populations. Bayesian clustering based on the eight nSSR loci demonstrated that the model with K = 2 explained the data satisfactorily (this model had the highest ∆K value). This finding suggested that the most probable number of populations was two based on our data for 849 individual from 28 naturally sampled populations. Cluster I comprised the LA, JX, AS, ZY and ZX populations, while cluster II comprised the remainder (Fig. 3). In order to identify any sub-clustering within each populations cluster, we separately ran new STRUCTURE analyses on the each cluster in the same manner as above. Repeating the structure analyses individually on clusters I and II resulted in highest ∆K value at K = 4 and K = 5, respectively. In cluster I, populations were split into four groups: (1) ZY, (2) AS, (3) ZX, (4) LA and JX while in cluster II, populations were split into five groups: (1) YX and SN, (2) LX, ZG, YC and RJ, (3) SH, CK, DC, and SX, (4) WD, CH, QL and YS, (5) WY, JS, YB, GM, FX, DB, ND, LB, and WL (Fig. 3). Similarly, the SAMOVA analysis also resulted in the highest F CT value (0.315) obtained when the 28 populations were divided into K = 2 groups: the ZY population and the other 27 populations.

Estimated clustering using STRUCTURE for K = 2 (a) based on nuclear mirosatellites and further analyses to determine substructure within the highest hierarchical level of genetic structure within cluster I for K = 4 (b) and cluster II for K = 5 (c) independently. Each individual is represented by a vertical line, divided into up to K colored segments representing the individual's estimated membership fraction to each of the K clusters. Vertical black lines separate samples from different locations as labeled on the bottom of the figure
For cpSSR data, the result of SAMOVA analysis showed that the highest F CT value (0.682) was obtained when the 25 populations were divided into K = 4 groups: ZY population, YX population, six populations composed of JS, QL, CK, YS, DC, SX, and the other 17 populations (Fig. 4). Based on bootstrap matrices, Monmonier's algorithm identified five boundaries defining zones of maximum genetic differences within the network of the 25 populations (Fig. 4). The most significant genetic barrier a was between CK population and GM population which separated most populations in Central China from other populations. Barrier b, c, and e around ZY population separated population ZY from other populations. Barrier d was around JX population which separated JX population from the others in Eastern China. Each of these boundaries was well-supported by the bootstrap analysis (with bootstrap values exceeding 50 %). The presence of these genetic barriers was further confirmed by analysis with single overall matrix (data not shown).

Genetic boundaries detected by Monmonier's algorithm based on chloroplast microsatellites. Black lines show Voronoı¨ tessellation and red lines show how Voronoı¨ tessellations act as boundaries. Those red lines with arrows at both ends indicate the five main boundaries (a, b, c, d and e) that are confirmed by analysis with single overall matrix and are all supported by high bootstrap values that are above 50 (data not shown). Symbols (dots, triangles, squares, and hexagons) show genetically different groups according to spatial analysis of molecular variance (SAMOVA) at K = 4
Mantel tests detected a significant correlation between genetic divergence, denoted by pairwise F ST/(1 − F ST) and geographical distances between populations for both nSSRs (r = 0.180, P < 0.013) and cpSSRs (r = 0.204, P < 0.001) (Fig. 5).

Isolation by distance analysis estimated using nSSRs (a) and cpSSRs (b). F ST/(1 − F ST) is plotted against the natural logarithms of the geographical distances (kilometers) between populations
Discussion
Genetic diversity and population bottleneck
A considerable high level of genetic diversity within C. mollissima populations as revealed by nSSRs (A = 7.4, A E = 3.8, A R = 5.4, H O = 0.590, H E = 0.678) and cpSSRs (h S = 0.433, h T = 0.895, v S = 0.278, v T = 0.902) might be explained by its wide geographic range. C. mollissima distributed in a large area of China, across sub-tropical, warm temperate and temperate regions, and in a large range of 50–2,800 m altitudes. Such vast regional and altitudinal range of C. mollissima should substantially contribute to the high level of genetic diversity detected in its populations.
C. mollissima populations from Central China were found to harbor the highest genetic diversity at both the nSSR and cpSSR loci, and in particular, GM population located in Shengnongjia Mountain (Hubei Province) exhibited the highest chloroplast haplotype diversity among all the examined populations. This result was in agreement with the study of Lang and Huang (1999), in which Shengnongjia area was pointed out to be the center of genetic diversity of the endemic Castanea species in China. Characterized by complex topographic features and climatic conditions, Central China has been recognized as one of the three plant diversity centers in China (Ying 2001). Warm climates experienced in this region during the Quaternary glaciation periods provided an ideal refuge center for many plants and animals, resulting in a diverse gene pool (Zheng 1983). Therefore, we inferred that the Central China, particularly the area around the Shengnongjia Mountain can be recognized as the modern center of genetic diversity of C. mollissima. In addition, the CK population sampled from the Dabashan Mountain natural reserve in Chengkou county (Chongqing city) has the highest number of private haplotype compared to all other populations. The Dabashan Mountain is characterized of diverse habitats, high species richness, and high endemism (Liu 2000; Xu 2003) and was recognized as one of prioritized regions for biodiversity conservation in China (Ministry of environmental protection 2011). The eastern part of the Dabashan Mountain borders with the Shengnongjia Mountain, where GM and CK population were sampled and has the highest haplotype diversity and highest number of private haplotype, respectively. In consideration of such a high variability in this region, we thus proposed that the Dabashan Mountains might be one refugium for C. mollissima. The same result has been reported by Wang et al. (2009) who recognized the Dabashan Mountains as one refugium for Eurycorymbus cavaleriei, a canopy tree in subtropical China. Furthermore, the chloroplast haplotype diversity in Southwestern China was just second to that in Central China, suggesting a high haplotype richness was also reserved in the region. As most sampled populations from Southwestern China were located in the Yunnan-Guizhou Plateau, the high genetic diversity in the region is well explained for the Yunnan-Guizhou Plateau, and its adjacent region have long been believed an important center of origin for the East Asiatic flora (Wang 1992) and were also identified as one of the three hot-spot for plant diversity in China (Ying 2001).
The F IS values were consistently positive in all populations and were significantly deviated from HWE in 18 populations (Table 2). This result could be ascribed to nonrandom mating promoted by population bottleneck. Although no significant deviation from mutation-drift equilibrium was detected under the SMM, signal of population reduction was identified in eight populations under the IAM (Table 1). Given that few microsatellite loci are expected to evolve strictly according to SMM, and the nSSRs examined in this study seemed to be IAM-like inferred from the allele distributions of the nSSR alleles (data not shown), so the bottleneck result based on IAM may be still informative. As the population size was reduced in bottlenecked populations, the frequency of inbreeding would be increased in those populations. In addition, several populations showed significant bottleneck are located in the marginal area of the species range (Fig. 1). The genetic exchange between those marginal populations with other populations would be less effective compared to populations located in central area, thus further promote inbreeding in those populations. Another explanation for the significant F IS values is probably due to the presence of null allele as it was detected for all the examined nSSRs. Therefore, the positive F IS values in C. mollissima may be attributable to population demographics (reduction of population size, inbreeding) and null alleles.
Genetic differentiation and population structure
The nSSRs revealed a relatively high level of genetic differentiation among C. mollissima populations (G′ ST = 0.447) compared to other species in Fagaceae, higher than Quercus crispula (G′ ST = 0.090, Ohsawa et al. 2011) and Castanopsis sclerophylla (G′ ST = 0.137, Wang et al. 2011). The G′ ST-based pollen-seed flow ratio of c. 3 in C. mollissima is much lower than that of other species in Fagaceae, for example Fagus sylvatica (60.6) and Q. suber (44.6) (Tsuda and Ide 2010). Those results suggested the pollen flow is not extensive and its contribution to migration is not so high in C. mollissima populations. The lack of extensive pollen flow was further confirmed by the results of STRUCTURE and SAMOVA analysis which both found that the most probable population clusters was 2, suggesting a clear genetic structure. However, populations from the same regions were not all assigned to the same groups which indicated that the distributions of genetic clusters were not strongly related to the geographical locations of the populations. Although AS, ZY, and ZX population in cluster I revealed by STRUCTURE analysis were geographically close to each other, they were split into three different groups as showed by the sub-clustering analysis on cluster I. The geographically discontinuous distribution of the clusters is probably due to the effect of human management. Because of its economic importance as nut tree crop, Chinese chestnut has a long history of cultivation in China. In order to pursue the economic benefits more rapidly, people once grafted the wild individuals with some superior cultivars to convert the wild populations into cultivated populations. Introduction of exotic cultivars which might lead to genetic exchange between wild populations and cultivated populations were also happened. The pronounced and long-term human involvement may have exerted substantial influence on the genetic structure of wild C. mollissima populations.
No signal of phylogeographic structure was found in the cpSSR data. Although pattern of isolation by distance was detected in the cpSSR, the linear regression explained only 4 % of the total variance (r 2 = 0.04). The seeds of C. mollissima are dispersed via gravity and animals. Stronger population genetic structure would be expected in a species with restricted seed dispersal (Duminil et al. 2007), so the possible effect of human activities should also be taken into account as a possible homogenizing factor. The nut of wild chestnut trees might been harvested and exchanged among different regions because of its economic importance as nut tree crop, thus promote the human-mediated seed dispersal. Little geographical structure of the genetic diversity was also found in C. sativa throughout southern European countries which was explained by the strong human impact on this species (Fineschi et al. 2000). Martín et al. (2012) also proposed that human influence to be the most plausible hypothesis for the origin of the southern populations of C. sativa in Spain given that there are no certain records of pollen in this area and their results highlight that the genetic structure of the current chestnut stands in Spain is the result of a natural colonization history and human-driven domestication.
Most pairwise values of F ST were significant, suggesting a possibility of adaptive divergence among natural populations of C. mollissima distributed across a wide geographic range in China. Local adaptation may have been evolved in such a large heterogeneous habitats and climatic conditions during long-term natural selection. An alternative explanation for the significant pairwise differentiation is probably due to genetic drift. The rate at which genetic drift alters allele frequencies depends on effective population size (Slatkin 1987). As some C. mollissima populations displayed signal of population reduction inferred from bottleneck analysis, the effect of genetic drift is also possible.
Landscape features may affect the gene flow and spatial genetic variation (Manel et al. 2003; Storfer et al. 2007). For instance, mountain ridges may provide geographical barriers that obstruct gene flow, thus producing genetic differentiation between populations across mountain ridges (Taberlet et al. 1998). In our study, three of the five identified barriers encircled ZY population, indicating a nearly complete isolation of ZY population from others and this result were also confirmed by SAMOVA analysis for both nSSRs and cpSSRs. ZY population in Zunyi city, Guizhou province was collected from a natural reserve which is located on the Daloushan Mountain. This natural reserve is characterized by complex topography and steep mountains, and the mountain ridges may acted as geographic barrier and thus disrupt the gene flow between ZY population and others. Such a strong isolation would lead to reduce of genetic variability and thus explained the lowest expected heterozygosity (H E = 0.600) observed in ZY population. The most significant genetic barrier between CK and GM population locates just where the Dabashan Mountain and Wushan Mountain situate. Dabashan Mountain locates in the border among Sichuan province, Hubei province and Shaanxi province, which extend in a northwest to southeast direction. Wushan Mountain situate in the common boundary between Hubei province and Sichuan province which extend in a northeast to southwest direction. The eastern parts of Dabashan Mountain adjoin Wushan Mountain and such a connection acts as a natural barrier. Barrier d was around JX population which was sampled from Qingliangfeng Mountain. The Qingliangfeng Mountain, which is the second highest mountain in eastern China, is located among Anhui province and Zhejiang province. Based on the above results, we propose that the mountain ridges have acted as significant barriers, causing the isolation of genetic lineages of C. mollissima. Similar conclusions have been drawn for a case study of Betula maximowicziana (Tsuda et al. 2010), that suggested mountain ridges acting as genetic barriers and restricting gene flow. In contrast, Magri et al. (2006) found that the mountain chains were not geographical barriers for beech (F. sylvatica) but rather than as a facilitated factor for its diffusion. This was probably due to the beech distribution feature in Europe where the beech did not cross the mountain tops (e.g., the Alps) but occurred along mountain slopes. However, the study of Fraxinus mandshurica conducted across five mountain ranges in Northeastern China found no clear effect of the mountains ridges acting as significant barriers (Hu et al. 2008). Thus, whether mountain ridges act as effective barriers to gene flow and migration or not will vary according to the species distribution patterns and modes of dispersal. The landscape genetic structure appear to be more complex than expected in the mountain ranges in China and information of the effects of such features need to be taken into account when designing programs to conserve and manage C. mollissima populations.
In this study, we investigated the geographical pattern of genetic diversity and population structure of natural C. mollissima populations in China at a large spatial scale using nuclear and chloroplast microsatellite markers for the first time. The acquired information should be conducive to formulating reasonable strategies for the conservation and utilization of wild resources of C. mollissima. The central China, particularly the area around the Shengnongjia Mountain, can be recognized as the modern center of genetic diversity of C. mollissima. In addition, high variability was also retained around the Dabashan Mountain. Therefore, wild populations of the species in those regions deserve prior conservation and utilization for breeding programs. Furthermore, adaptive differentiation might be evolved among populations thus core germplasm repository should be constructed through representative sampling of C. mollissima from the different eco-regions of the species distribution range. In addition, genetically isolated populations such as ZY population because of the mountain ridges blocking gene flow should also be prioritized when formulating in situ and ex situ conservation strategies for C. mollissima. In the present study, we utilized nuclear and chloroplast microsatellite markers which are generally considered to be selection-neutral and thus fail to detect genetic variations associated with natural selection or adaptive evolution. With increasing genomic resources for Chinese chestnut (www.fagaceae.org), future works should focus on exploring the genetic variations related to adaptive traits using whole-genome scan based on functional markers such as EST-SSR and EST-SNP.
References
Anagnostakis SL (1992) Measuring resistance of chestnut trees to chestnut blight. Can J For Res 22:568–571
Barakat A, DiLoreto D, Zhang Y, Smith C, Baier K, Powell W, Wheeler N, Sederoff R, Carlson J (2009) Comparison of the transcriptomes of American chestnut (Castanea dentata) and Chinese chestnut (Castanea mollissima) in response to the chestnut blight infection. BMC Plant Biol 9:51
Bohonak A (2002) IBD (isolation by distance): a program for analyses of isolation by distance. J Hered 93:153–154
Burnham CR (1988) The restoration of the American chestnut: Mendelian genetics may solve a problem that has resisted other approaches. Am Sci 76:478–487
Chapuis MP, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24:621–631
Crawford NG (2010) SMOGD: software for the measurement of genetic diversity. Mol Ecol Resour 10:556–557
Crow JF (2002) Perspective: here's to Fisher, additive genetic variance, and the fundamental theorem of natural selection. Evolution 56:1313–1316
Dane F, Lang P, Huang HW, Fu Y (2003) Intercontinental genetic divergence of Castanea species in eastern Asia and eastern North America. Heredity 91:314–321
Deguilloux MF, Dumolin-Lapègue S, Gielly L, Grivet D, Petit R (2003) A set of primers for the amplification of chloroplast microsatellites in Quercus. Mol Ecol Notes 3:24–27
Dieringer D, Schlötterer C (2003) Microsatellite analyser (MSA): a platform independent analysis tool for large microsatellite data sets. Mol Ecol Notes 3:167–169
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem bull 19:11–15
Duminil J, Fineschi S, Hampe A, Jordano P, Salvini D, Vendramin GG, Petit RJ (2007) Can population genetic structure be predicted from life-history traits? Am Nat 169:662–672
Dupanloup I, Schneider S, Excoffier L (2002) A simulated annealing approach to define the genetic structure of populations. Mol Ecol 11:2571–2581
El Mousadik A, Petit R (1996) Chloroplast DNA phylogeography of the argan tree of Morocco. Mol Ecol 5:547–555
Ennos R (1994) Estimating the relative rates of pollen and seed migration among plant populations. Heredity 72:250–259
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Fineschi S, Taurchini D, Villani F, Vendramin G (2000) Chloroplast DNA polymorphism reveals little geographical structure in Castanea sativa Mill. (Fagaceae) throughout southern European countries. Mol Ecol 9:1495–1503
Fontaine C, Lovett P, Sanou H, Maley J, Bouvet J (2004) Genetic diversity of the shea tree (Vitellaria paradoxa CF Gaertn), detected by RAPD and chloroplast microsatellite markers. Heredity 93:639–648
Goodman SJ (1997) R ST Calc: a collection of computer programs for calculating estimates of genetic differentiation from microsatellite data and determining their significance. Mol Ecol 6:881–885
Goudet J (2001) FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9. 3). Available from http://www2.unil.ch/popgen/softwares/fstat.htm
Hardy OJ, Vekemans X (2002) SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2:618–620
Hedrick PW (2005) A standardized genetic differentiation measure. Evolution 59:1633–1638
Hu LJ, Uchiyama K, Shen HL, Saito Y, Tsuda Y, Ide Y (2008) Nuclear DNA microsatellites reveal genetic variation but a lack of phylogeographical structure in an endangered species, Fraxinus mandshurica, across north-east China. Ann Bot 102:195–205
Huang HW (1998) Review of current research of the world Castanea species and importance of germplasm conservation of China native Castanea species. J Wuhan Bot Res 16:171–176
Huang HW, Dane F, Kubisiak TL (1998) Allozyme and RAPD analysis of the genetic diversity and geographic variation in wild populations of the American chestnut (Fagaceae). Am J Bot 85:1013–1021
Huang HW, Dane F, Norton JD (1994) Allozyme diversity in Chinese, Seguin and American chestnut (Castanea spp.). Theor Appl Genet 88:981–985
Jump AS, Penuelas J (2005) Running to stand still: adaptation and the response of plants to rapid climate change. Ecol Lett 8:1010–1020
Kubisiak TL, Hebard F, Nelson CD, Zhang J, Bernatzky R, Huang HW, Anagnostakis S, Doudrick R (1997) Molecular mapping of resistance to blight in an interspecific cross in the genus Castanea. Phytopathology 87:751–759
Lang P, Huang HW (1999) Genetic diversity and geographic variation in natural populations of the endemic Castanea species in China. Acta Bot Sin 41:651–657
Liu YC (2000) Flora geography of national wild conservative plants in Chongqiong. J Southwest China Norm Univ (Nat Sci) 25:439–447
Luikart G, Cornuet JM (1998) Empirical evaluation of a test for identifying recently bottlenecked populations from allele frequency data. Conserv Biol 12:228–237
Magri D, Vendramin GG, Comps B, Dupanloup I, Geburek T, Gömöry D, Latałowa M, Litt T, Paule L, Roure JM (2006) A new scenario for the Quaternary history of European beech populations: palaeobotanical evidence and genetic consequences. New Phytol 171:199–221
Manel S, Schwartz MK, Luikart G, Taberlet P (2003) Landscape genetics: combining landscape ecology and population genetics. Trends Ecol Evol 18:189–197
Manni F, Guerard E, Heyer E (2004) Geographic patterns of (genetic, morphologic, linguistic) variation: how barriers can be detected by using Monmonier's algorithm. Hum Biol 76:173–190
Mantel N (1967) The detection of disease clustering and a generalized regression approach. Cancer Res 27:209–220
Marinoni D, Akkak A, Bounous G, Edwards KJ, Botta R (2003) Development and characterization of microsatellite markers in Castanea sativa (Mill.). Mol Breeding 11:127–136
Martín MA, Mattioni C, Molina JR, Alvarez JB, Cherubini M, Herrera MA, Villani F, Martín LM (2012) Landscape genetic structure of chestnut (Castanea sativa Mill.) in Spain. Tree Genet Genomes 8:127–136
Martin MA, Mattioni C, Cherubini M, Taurchini D, Villani F (2010) Genetic diversity in European chestnut populations by means of genomic and genic microsatellite markers. Tree Genet Genomes 6:735–744
Martins K, Chaves L, Vencovsky R, Kageyama P (2011) Genetic structure based on nuclear and chloroplast microsatellite loci of Solanum lycocarpum A. St. Hil. (Solanaceae) in Central Brazil. Genet Mol Res 10:665–677
Mattioni C, Cherubini M, Micheli E, Villani F, Bucci G (2008) Role of domestication in shaping Castanea sativa genetic variation in Europe. Tree Genet Genomes 4:563–574
Monmonier MS (1973) Maximum-difference barriers: an alternative numerical regionalization method*. Geogr Anal 5:245–261
Ministry of environmental protection (2011) China national biodiversity conservation strategy and action plan. China Environmental Science Press, Beijing
Nei M (1987) Molecular Evolutionary Genetics. Columbia University Press, New York
Nei M, Tajima F, Tateno Y (1983) Accuracy of estimated phylogenetic trees from molecular data. J Mol Evol 19:153–170
Newton A, Allnutt T, Dvorak W, Del Castillo R, Ennos RA (2002) Patterns of genetic variation in Pinus chiapensis, a threatened Mexican pine, detected by RAPD and mitochondrial DNA RFLP markers. Heredity 89:191–198
Ohsawa T, Tsuda Y, Saito Y, Ide Y (2011) The genetic structure of Quercus crispula in northeastern Japan as revealed by nuclear simple sequence repeat loci. J Plant Res 124:645–654
Parker IM, Gilbert GS (2004) The evolutionary ecology of novel plant-pathogen interactions. Annu Rev Ecol Evol Syst 35:675–700
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Petit RJ, El Mousadik A, Pons O (1998) Identifying populations for conservation on the basis of genetic markers. Conserv Biol 12:844–855
Petit RJ, Duminil J, Fineschi S, Hampe A, Salvini D, Vendramin GG (2005) Comparative organization of chloroplast, mitochondrial and nuclear diversity in plant populations. Mol Ecol 14:689–701
Piry S, Luikart G, Cornuet J-M (1999) Computer note. BOTTLENECK: a computer program for detecting recent reductions in the effective size using allele frequency data. J Hered 90:502–503
Pons O, Petit R (1996) Measuring and testing genetic differentiation with ordered versus unordered alleles. Genetics 144:1237–1245
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Sanguinetti C, Dias NE, Simpson A (1994) Rapid silver staining and recovery of PCR products separated on polyacrylamide gels. Biotechniques 17:914–921
Sebastiani F, Carnevale S, Vendramin G (2004) A new set of mono-and dinucleotide chloroplast microsatellites in Fagaceae. Mol Ecol Notes 4:259–261
Slatkin M (1987) Gene flow and the geographic structure of natural populations. Science 236:787–792
Storfer A, Murphy M, Evans J, Goldberg C, Robinson S, Spear S, Dezzani R, Delmelle E, Vierling L, Waits L (2007) Putting the ‘landscape’in landscape genetics. Heredity 98:128–142
Taberlet P, Fumagalli L, Wust-Saucy AG, Cosson JF (1998) Comparative phylogeography and postglacial colonization routes in Europe. Mol Ecol 7:453–464
Takahashi T, Tani N, Niiyama K, Yoshida S, Taira H, Tsumura Y (2008) Genetic succession and spatial genetic structure in a natural old growth Cryptomeria japonica forest revealed by nuclear and chloroplast microsatellite markers. Forest Ecol Manag 255:2820–2828
Tanaka T, Yamamoto T, Suzuki M (2005) Genetic diversity of Castanea crenata in northern Japan assessed by SSR markers. Breeding Sci 55:271–277
Tsuda Y, Ide Y (2010) Chloroplast DNA phylogeography of Betula maximowicziana, a long-lived pioneer tree species and noble hardwood in Japan. J Plant Res 123:343–353
Tsuda Y, Sawada H, Ohsawa T, Nakao K, Nishikawa H, Ide Y (2010) Landscape genetic structure of Betula maximowicziana in the Chichibu mountain range, central Japan. Tree Genet Genomes 6:377–387
Villani F, Pigliucci M, Benedettelli S, Cherubini M (1991) Genetic differentiation among Turkish chestnut (Castanea sativa Mill.) populations. Heredity 66:131–136
Wang J, Gao PX, Kang M, Lowe AJ, Huang HW (2009) Refugia within refugia: the case study of a canopy tree (Eurycorymbus cavaleriei) in subtropical China. J Biogeogr 36:2156–2164
Wang R, Compton SG, Chen X-Y (2011) Fragmentation can increase spatial genetic structure without decreasing pollen-mediated gene flow in a wind-pollinated tree. Mol Ecol 20:4421–4432
Wang WT (1992) On some distribution patterns and some migration routes found in the eastern Asiatic region. Acta Phytotaxon Sin 30:1–24
Wang Y, Kang M, Huang HW (2008) Microsatellite loci transferability in chestnut. J Am Soc Hort Sci 133:692–700
Wheeler N, Sederoff R (2009) Role of genomics in the potential restoration of the American chestnut. Tree Genet Genomes 5:181–187
Xu DY (2003) An analysis of seed plants floral composition in Dabashan Nature Reserve. J Southwest China Norm Univ (Nat Sci) 28:963–968
Yamamoto T, Tanaka T, Kotobuki K, Matsuta N, Suzuki M, Hayashi T (2003) Characterization of simple sequence repeats in Japanese chestnut. J Hortic Sci Biotech 78:197–203
Ying JS (2001) Genetic diversity and distribution pattern of seed plants in China. Biodiv Sci 9:393–398
Zheng Z (1983) The characteristics of the flora and an outline of the distribution of plants in Hubei Province, People's Republic of China. J Wuhan Bot Res 1:165–175
Acknowledgments
We thank two anonymous reviewers for their valuable suggestions and critical comments on the earlier version of the manuscript. This project was funded by the National Science Foundation of China (30771479) and key initiative grant of the Chinese Academy of Sciences (KSCX2-YW-N-061).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by A. Kremer
W. Liu and M. Kang contributed equally to this work.
Rights and permissions
About this article
Cite this article
Liu, W., Kang, M., Tian, H. et al. A range wide geographic pattern of genetic diversity and population structure of Castanea mollissima populations inferred from nuclear and chloroplast microsatellites. Tree Genetics & Genomes 9, 975–987 (2013). https://doi.org/10.1007/s11295-013-0610-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11295-013-0610-3