
Abstract
The fungus Slafractonia leguminicola, the causal agent of blackpatch disease of legumes produces two mycotoxins slaframine and swainsonine, causing slobbers’ symptoms and locoism of grazing animals, respectively. The genetics of this important fungus is poorly understood. This work aimed to develop a genetic transformation system and evaluate the efficacy of RNA interference (RNAi) in S. leguminicola. In this study, S. leguminicola was transformed using a PEG-mediated method with a fungal construct that carries a hygromycin resistance cassette. To assess the use of RNAi, a silencing construct pSilentPKS1-AS was constructed which includes inverted repeat transgenes of the polyketide synthase gene (pks1) that is involved in melanin biosynthesis. Transformation of S. leguminicola with the IRT pks1 vector decreased pks1 transcripts levels 82–92% in knockdown mutants when compared with the wild type and was accompanied with a reduction in melanin and swainsonine production. These results demonstrate that RNAi can be a useful tool for studying gene function in S. leguminicola.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The fungal plant pathogen Slafractonia leguminicola (Gough & E.S. Elliot) Alhawatema, Baucom, Sanogo & Creamer (formerly Rhizoctonia leguminicola) (Alhawatema et al. 2015), causes “slobbers”, a toxicosis of livestock that graze in fields infested with S. leguminicola (Crump et al. 1963; Hagler and Croom 1989; Smalley et al. 1962). Symptoms of slobbers include excessive salivation, diarrhea, frequent urination, loss of appetite, and in severe cases even death, that become apparent after animals consume hosts, such as clover infected with S. leguminicola for several days (Aust 1974; Aust et al. 1966). The mycotoxin slaframine, an indolizidine alkaloid that is produced by S. leguminicola induces slobbers (Croom et al. 1995).
In addition to slaframine, S. leguminicola produces another chemically similar alkaloid mycotoxin, swainsonine, that induces a different toxicosis, locoism, in livestock animals (Croom et al. 1995). Swainsonine is also produced by the Alternaria section Undifilum fungal endophytes of Astragalus sp. and Oxytropis sp. locoweed plants (Braun et al. 2003). Both swainsonine and slaframine are synthesized from a common intermediate, pipecolic acid, which can be derived from l-lysine (Guengerich and Broquist 1973; Guengerich et al. 1973; Wickwire et al. 1990). Recent work showed that all swainsonine-producing fungi, including S. leguminicola, contain a polyketide synthase gene cluster, swnK, shown to be essential for swainsonine biosynthesis (Cook et al. 2017). For S. leguminicola, swnK, and the associated genes, swnN, swnR, and swnT were located on separate scaffolds, while swnH1 and swnH2 were located on the same scaffold. This fungus also contained swnK paralogs with only 54% identity with the S. leguminicola swnK, which presumably are involved in slaframine biosynthesis.
Slafractonia leguminicola was previously misidentified as Rhizoctonia leguminicola. Through comparisons of morphology and molecular phylogenetics of the glyceraldehyde-3-phosphate dehydrogenase (gpd) and internal transcribed spacer region (ITS) with other fungi, the fungus was shown to be an ascomycete (Alhawatema et al. 2015).
RNA silencing has been used to investigate gene function in toxin-producing fungi (Bdel-Hadi et al. 2011). RNA interference (RNAi) is the common process of gene regulation that utilizes double stranded RNA molecules (dsRNAs) to induce the degradation of specific mRNA species (Salame et al. 2011). The RNAi is activated in the cell when the ribonuclease Dicer binds and cleaves dsRNAs to produce fragments of 20–25 bp in length. These fragments, called small interfering RNAs (siRNAs), are then separated into single strands and integrated into the RNA induced silencing complex (RISC), which binds and cleave similar mRNAs and thus decreases gene expression (Ahlquist 2002; Bernstein et al. 2001; Zamore et al. 2000). Down regulation of gene expression by RNAi has been successfully demonstrated in several fungal species such as Aspergillus fumigatus, Bipolaris oryzae, Colletotrichum lagenarium, and Cryptococcus neoformans (Bdel-Hadi et al. 2011; Liu et al. 2002; Mouyna et al. 2004; Nakayashiki et al. 2005; Salame et al. 2011). This technique in ascomycetes has been initiated by genetic transformation with fungal vectors that contain inverted repeat transgenes (IRT) separated by a fungal intron and a hairpin dsRNA structure (Akihiro et al. 2007; Liu et al. 2002; Mouyna et al. 2004; Nakayashiki et al. 2005). Transformation of the fungal species B. oryzae and M. grisea with IRT containing homologous sequences to the polyketide synthase gene (pks1), that is involved in the melanin pigment, generated knockdown mutants with a range of 70–100% silencing and verified that RNAi could be highly efficacious in fungi (Akihiro et al. 2007; Nakayashiki et al. 2005).
Although S. leguminicola causes economic damage to grazing animals in infested fields (Croom et al. 1995; Gough and Elliott 1956), little information exists on the genetics of the fungus. This is in part because of the lack of developed genetic tools. This study sought to develop genetic tools, including a transformation system and an RNAi system, to study gene function in S. leguminicola. Since the pks1 gene is responsible for melanin production and thus the dark color in many fungi, successful silencing using RNAi would result in light to white phenotypes. Results obtained by this easily observable trait could be used to demonstrate the applicability of RNAi to suppress endogenous genes in S. leguminicola.
Materials and methods
Fungal strains
The S. leguminicola isolate (RL-4038), originally isolated from red clover and obtained from the American Type Culture Collection (ATCC), was used in transformation experiments. The fungus was maintained on PDA at 26 °C.
RNA extraction and cDNA synthesis
Total RNA was extracted from S. leguminicola mycelia according to the manufacturer’s protocol using an RNeasy mini Kit (Qiagen, Valencia, CA, USA). The RNA concentration was measured using the Nano Drop (Thermo Fisher Scientific, Wilmington, DE, USA). A 400 ng aliquot of S. leguminicola RNA was used to produce first strand cDNA using the SuperScript® III First-Strand cDNA Synthesis System (Invitrogen, CA., USA).
Identification of pks1 melanin gene
To identify the melanin coding sequence of the pks1 gene, several primers were designed (Table 1. PSF2, PSF6, and PSR3) based on the alignment of melanin pks 1 sequences from related fungi including Alternaria alternata pks1, (GenBank# HM486910.1) and Phoma pinodella pks1, (GenBank# GQ150547.1). The PCR protocol used to amplify the pks1 fragments from S. leguminicola cDNA was as follows: an initial denaturation at 95 °C for 2 min followed by 35 cycles of 1 min at 95 °C, 1 min at 57 °C, 2 min at 72 °C and a final extension for 5 min. All reactions were carried out in 50 µL volume and each contained 2 mM MgCl2, 0.3 µm of each primer, 400 µm dNTPs, 0.5 µL of GoTaq, and 10 µL of 5 × buffer (Promega, Madison, WI, USA). PCR products were purified using QIAquick PCR Purification Kit (Qiagen, Valencia, CA., USA) and sequenced at Sequetech Corporation (CA, USA). For each sequence obtained, sequence similarity matches were determined using BLASTn.
Transformation vectors
The fungal transformation vector pSilent-1 (Nakayashiki et al. 2005) of approximately 7.0 kB was used in silencing experiments. Transformants were selected based on the antibiotic hygromycin B (hyg B) since the fungal pSilent-1 vector confers resistance against hyg B to transformants. We used the pSilent-1 vector to construct a silencing vector for the pks1 gene. The strain JM109 of E. coli was used for the maintenance and propagation of transformation vectors throughout this study. Plasmid DNA was extracted and purified using the manufacturer’s protocols for a plasmid mini kit (Qiagen, Valencia, CA., USA).
Plasmid construction of the pks1 vector pSilentPKS1-AS
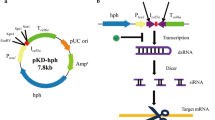
Vector pSilent-1 (Nakayashiki et al. 2005) was used with inverted repeats of a coding pks1 gene segment (IRT pks1) (480 bp) to build the silencing vector for pks1, pSilentPKSi-AS (Fig. 1). The pSilentPKS1-AS was built in two steps (Akihiro et al. 2007). The pks1 fragments (480 bp) were amplified by PCR from cDNA of S. leguminicola using PCR primers (PSF2-Xhol or PSF6-Xhol and PSR3-HindIII) (Table 1). These amplified fragments of pks1 were digested with the restriction enzymes XhoI and HindIII, and cloned into the XhoI-HindIII site of the pSilent-1 vector, producing pSilentPKS1-S (Fig. 1). Another pks1 fragment was amplified using PCR primers (PSF2- SphI or PSF6- SphI and PSR3-BglII) (Table 1). Once digested, the fragment was cloned in an inverted direction into the SphI–BglII site to form the IRT pks1 vector pSilentPKS1-AS (Fig. 1).

Schematic representations of the silencing construct pSilentPKS1-AS encoding pks1 hairpin. Arrows in the construct indicate the direction of the pks1coding region. IT, intron 2 of the cutinase (CUT) gene from Magnaporthe oryzae; PtrpC, Aspergillus nidulans trpC promotor; TtrpC, A. nidulans trpC terminator; hyg B, hygromycin resistant gene; Amp, ampicillin resistant gene (Nakayashiki et al. 2005)
Preparation of protoplasts
Four plugs of 4 mm diameter were cut using a sterilized cork borer from the advancing hyphae of 5-day-old S. leguminicola cultures and were suspended in 4 mL of Czapek-Dox broth medium (CDB, Difco, USA). After grinding four fungal plugs using mortar and pestle, 1.5 mL of suspension was transferred into 2 mL microfuge tubes, and the suspension was pelleted by centrifugation at 13,000 rpm for 10 min at room temperature. The fungal pellets were transferred to a 15 mL centrifuge tube containing 13 mL of 0.9 M MgSO4 of 14 mg/mL lysing enzyme (L1412, Sigma, USA) and the suspension was incubated for 4.5 h at room temperature with 50 rpm shaking. The suspension was then filtered through two layers of lens cleaning tissues to separate most mycelium residues from protoplasts. The filtered protoplasts were then pelleted by centrifugation at 600×g for 10 min at 4C°. The supernatant was discarded and the protoplasts were washed with 6 mL 0.6 M KCl, 50 mM CaCl2 and pelleted by centrifugation at 600×g for 10 min at 4° C. The pelleted protoplasts were suspended in 300 mL of 0.6 M KCl, 50 mM CaCl2 buffer and kept on ice.
PEG mediated DNA transformation
Fresh protoplasts 100 μL in stabilizer buffer (0.6M KCl, 50 mM CaCl2) were mixed with 1–10 μg of plasmid pSilent-1 (control, empty vector) or pSilentPKS1-AS (Fig. 3) in separate 15 mL centrifuge tubes and incubated for 10 min on ice. Then, 50 μL of a polyethylene glycol 3350 (PEG) buffer (25% PEG, 50 mM CaCl2, 10 mM Tris–HCl) was added to the protoplast suspension with gentle shaking. After 20 min on ice, another 250 μL of the PEG buffer was added to the protoplast suspension and the mixture was incubated for 10 min on ice. The mixture was then incubated at room temperature for 20 min. Then another 3 mL of PEG buffer was added and the mixture was incubated at room temperature for another 20 min. The protoplast transformation mixture was poured (500 μL/plate) onto several plates that already contained 15 mL of cold regeneration medium (6 M sucrose, 0.3% (w/v) yeast extract, 0.3% (w/v) casamino acids, and 1.6% (w/v) agar). The plates containing transformed protoplasts were gently shaken by hand and incubated at 29 °C. After 48 h incubation, the plates were layered with 10 mL of the regeneration medium containing 70 μL hyg B /mL and incubated for 7–14 days until the appearance of colonies. Colonies that appeared within 7–14 days on hyg B—containing plates were directly transferred to PDA to study their colony phenotype.
Transformants verification by PCR
To confirm that transformants contained the silencing vector, a 300 bp segment of the hyg B gene was amplified and sequenced from selected transformants by PCR using specific primers cdH2 and cdH3 (Table 1).
Real-time reverse transcription quantitative PCR (RT-qPCR)
To check the expression pattern of six selected IRT pks1 transformants, a wild type isolate, and a wild type isolate transformed with EV (empty vector, pSilent-1), RT-PCR was performed. After growing the IRT pks1 transformants on PDA for five days, total RNA was extracted and quantified as described above. A 4 μg aliquot of total RNA from each sample was used to produce 20 μL first strand cDNA using SuperScript® III First-Strand System cDNA synthesis (Invitrogen, CA., USA). A 15 μL aliquot of cDNA from transformants and wild type was diluted by a factor of 140 with nuclease-free water to get a concentration of 10 ng per 7 μL. Seven microliters of the dilution containing 10 ng were used in the RT-PCR reactions.
GPDH, PGK1, RDN5.8, and UBC7 (Table 2) were used as internal controls for comparison to the target gene pks1 as described in a study of fungal reference genes for RT-PCR (Li et al. 2012). The pks1-qPCR primers were designed from the pks1 sequence (sense strand) of pSilentPKS1-AS using the software PrimerQuest of Integrated DNA Technologies (http://idtdna.com).
Quantitative PCR reactions were done using the Bio Rad CFX96 platform (Bio Rad, Hercules, CA, USA) with the SsoAdvanced Universal SYBER GREEN Supermix kit (Bio Rad, Hercules, CA, USA). The RT-PCR program was 95 °C for 3 min, followed by 40 cycles of 95 °C for 10 s, 58 °C for 30 s, followed by a melting curve analysis. All reactions were carried out in 20 µL volumes and each contained 10 µL of SsoAdvanced Universal SYBER GREEN Supermix (2X), 7 µL cDNA (10 ng), and 3 µL (300 nM) of forward and reverse primers. All tests were carried out with three technical replicates and data were analyzed using CFX manager software. The relative normalized gene expression was calculated using the ΔΔ Cq method.
To study the primers efficiency during the amplification, standard curves were made from six ten fold dilutions. Five microliters of cDNA containing (1000 ng) were taken from each sample and pooled. The solution was diluted to 1:14 with sdH2O to get a concentration of 100 ng per 7 μL. Six ten fold dilutions were made from this stock solution (100–0.001 ng). The reactions were carried out as previously mentioned. Efficiency parameters such efficiency percentage (E) and standard curve regression coefficient (R2) were generated by Bio Rad CFX96 software manager. Efficiency percentage of (90–110%) and R2 = 0.98 are considered acceptable values for primer amplification efficiency. To ensure that the increase in fluorescence resulting from SYBR green binding to DNA was specific to primer amplification, melting curves were generated at the end of the run with small increments in temperature (0.05 °C).
Swainsonine analysis
Fungal cultures of S. leguminicola: wild type, wild type transformed with EV (empty vector, pSilent-1), and three selected pks1 mutants, were grown on PDA plates for eight days at 28 °C. The fungal mass of each culture was dried at 80 °C for 3 h. Dry mycelia (100 mg) from each culture were accurately weighed and extracted for swainsonine according to a previously published method (Gardner and Cook 2010). Liquid chromatography–Mass spectrometry (LC–MS) analysis of samples was done at the USDA Poisonous Plant Research Laboratory, Logan, UT. Swainsonine levels were compared using paired Student t test analyses.
Results
Blast Analysis of pks1 melanin gene
Using RT-PCR (reverse transcriptase PCR) and cDNA as template, amplicons of 530 or 400 bp were obtained utilizing PKSF2 and PKSR3 or PKSF6 and PKSR3 primers, respectively (Table 1). Using BLASTn, the pks1 sequence (GenBank # LC146404.1) showed 85% identity with sequences from Alternaria alternata ALM and 82% with Ascochyta rabiei polyketide synthesis genes.
Transformation of S. leguminicola
Protoplasts of S. leguminicola were transformed with the IRT pks1 vector pSilentPKS1-AS, and the vector pSilent-1 as a control. We used fresh protoplasts released after a 4.5-h treatment of young hyphae with lysing enzyme (L1412, Sigma, USA). After 7–14 days of incubation, a number of separate fungal hyg B resistant colonies appeared on the selective regeneration plates. Using pSilent-1 and pSilentPKS1-AS plasmids, the efficacy of transformation was low, 0.5–1 resistant colonies per µg DNA.
Screening of IRT pks1 transformant morphology
Slafractonia leguminicola protoplasts were transformed with the pSilentPKS1-AS construct using PEG -mediated transformation as previously described. Colonies that appeared within 7–14 days on hyg B resistance plates were directly transferred to PDA.
Five days after transferring IRT pks1 transformants to PDA, transformants segregated into two phenotypic shapes (Fig. 2). One group included pks1 transformants for which the colony had an irregular shape and white mycelia (close to albino color) (Fig. 2, T1 and T6). The second group included pks1 transformants that had a regular circular phenotype with light to white mycelia color (Fig. 2, T2–T5). The pSilent-1 transformants (empty plasmid, EV) had the same phenotypic appearance of the wild type (Fig. 2, EV).

Colony phenotypes of Slafractonia leguminicola wild type and IRT pks1 transformants. Wt wild type S. leguminicola and E V: wild-type S. leguminicola transformed with empty vector (pSilent-1) after 3 days of growth on PDA, IRT pks1 transformants (T1–T6) after 5 days of growth on PDA after transformation with the silencing vector pSilentPKS1-AS
Transformant verification by PCR
The IRT pks1 transformants (T1–T6), the wild type, and the wild type transformed with pSilent-1 (E V) were tested by PCR using primers cdH2 and cdH3 (Table 1) to determine if the transformants harbored the hyg B gene. Only transformants (T1–T6 and EV) showed the expected hyg B gene band (Fig. 3) and BLASTn results showed 99% identity to pSilent-1 (GenBank # AB303070.2).

Agarose gel electrophoresis of wild type and hygromycin-resistant transformants tested by PCR for presence of the transgene hyg B segment (300 bp). Wt wild-type S. leguminicola, E V wild-type S. leguminicola transformed with the empty vector (pSilent-1), pks1 transformants (T1–T6) after 5 days growth on PDA after transformation with the silencing vector pSilentPKS1-AS
RT-PCR
Quantitative PCR was performed to confirm that the reduction in melanin pigment in IRT pks1 transformants was due to reduced levels of pks1 mRNA.
The GPDH, PGK1, and UBC7 primers had low amplification efficiency, since they were not consistently amplified at different dilutions and gave non-specific amplification. Therefore, we used only RDN5.8 to normalize the results for the target gene pks1. The linear correlation coefficient (R2) for the reference gene RDN5.8 and the target gene pks1 ranged from 0.99 to 1.0. Based on these slopes for the standard curves of both genes (data not shown), the amplification efficiencies of the primers were within the accepted range (90–110%). Melting curves of pks1 and RDN5.8 primers were generated at the end of run in order to check for the presence of non-specific amplifications. The existence of more than one peak is indicative of non-specific amplification or primer dimer. The melting curves revealed one peak for each primer set (data not shown).
Quantitative PCR results indicated that IRT pks1 transformants with light to white mycelia showed a decrease in pks1 mRNA levels compared with controls (Fig. 4). The pks1 gene was strongly expressed in the wild type (Wt) and the wild type transformed with the empty vector E V (pSilent-1) compared with all IRT pks1 transformants, T1 through T6 (Figs. 2, 4). Of the IRT pks1 transformants tested, T2, and T4 showed an 82% reduction in pks1 relative expression level compared with Wt, while T3 showed the highest decrease in pks1 expression (92%) compared with Wt (Fig. 4).

Quantitative real time PCR analysis of total RNA from wild type, and IRT pks1 transformants. RNA was extracted, converted to cDNA, and the expression of pks1 was tested. The expression pattern of IRT pks1 was normalized to the expression pattern of RDN5.8 gene in each sample. Samples were blotted against relative normalized expression. Wt wild-type S. leguminicola, E V wild-type S. leguminicola transformed with empty vector (pSilent-1), IRT pks1 transformants (T1–T6) of 5 days of growth on PDA after transformation with the silencing vector pSilentPKS1-AS (Fig. 2). Error bars represent the standard error of the mean (n = 3)
Swainsonine analysis
There were highly statistically significant reductions in swainsonine concentrations between the three IRT pks1 transformants (T2, T3, and T4) compared with Wt (Figs. 5, 6). In contrast, there was no significant reduction in swainsonine concentration between the wild type and the wild type transformed with EV (Psilent-1, empty vector). Swainsonine concentration in mycelia dry weight was decreased to 43% in T2, 46% in T3, and to 48% in T4 (Fig. 6).

Colony phenotypes of S. leguminicola wild type and IRT pks1 transformants after 8 days of growing on PDA after transformation with pSilentPKS1-AS. Wt wild-type S. leguminicola, E V wild-type S. leguminicola transformed with empty vector (pSilent-1), IRT pks1 transformants (T2–T4) of 8 days of growing on PDA after transformation with the silencing vector pSilentPKS1-AS

Comparison of swainsonine (SW) concentrations (%) in dry weight (d.w.) of mycelia between the three IRT pks1 transformants (T2, T3, and T4), the wild type Wt and the wild type transformed with EV (Psilent-1, empty vector). Significant difference with wild type (Wt) were evaluated by t test and shown by asterisks (P** < 0·01). Error bars represent the standard error of the mean (n = 3)
Discussion
This is the first study to report genetic transformation and to demonstrate the use of RNAi for genetic manipulation of S. leguminicola. We demonstrated transformation of S. leguminicola with hyg B gene by PEG- mediated DNA transformation. It was not surprising to achieve low transformation rates since PEG-mediated transformation has been reported to provide low efficacy rate (O’Neill et al. 1998).
The PKS of S. leguminicola was most closely related to Alternaria alternata ALM and Ascochyta rabiei polyketide synthesis genes, both of which have been reported to play a role in melanin biosynthesis (Akamatsu et al. 2010; Kawamura et al. 1997). Our results confirmed that the study was carried out with a portion of the gene associated with melanin biosynthesis. However, since the ketide synthase active site is highly conserved across many fungi, this likely contributed to off-target silencing effects.
To study RNAi interference in S. leguminicola, the IRT pks1 silencing vector, pSilentPKS1-AS, was designed from pSilent-1 by two cloning steps. Transformation of S. leguminicola with pSilentPKS1-AS vector resulted in phenotypes that were close to albino at early stages of growth to light grey color at later stages of hyphal growth. Unlike S. leguminicola, knockdowns of pks1 gene of C. neoformans (Liu et al. 2002), (A) fumigatus (Mouyna et al. 2004), and (B) oryzae (Akihiro et al. 2007) resulted in complete RNAi albino phenotypes. However, we never observed 100% silencing of pks1 transcripts and the complete albino phenotype was not observed for IRT pks1 transformants. This could be due to different integrations of the silencing vector in the fungal genome resulting in varied expressions of the RNAi construct (Mouyna et al. 2004).
Assessment of the pks1 mRNA levels among IRT pks1 transformants and the wild type S. leguminicola using RT-qPCR indicated there were significant reductions of pks1 transcript levels in IRT pks1 transformants when compared with control samples. When comparing the same IRT pks1 transformants after 8 days of growing on PDA with 5 days of growing on PDA, it was clear that IRT pks1 transformants retain some ability to produce melanin pigment over time, and all IRT pks1 transformants had a light grey color after 8 days. This suggests some level of melanin production, despite the significant decrease in pks1 transcript levels found during early growth. The gene silencing could have been partially suppressed in the fungus in days 5–8, implying that RNAi in S. leguminicola was not efficacious over time and would be better used for studying transient gene function in S. leguminicola.
Swainsonine analysis showed that a decrease in pks1 expression was followed by a significant reduction in swainsonine concentrations in IRT pks1 transformants in relation to controls. One reason for this significant decrease in swainsonine amount could be due to off-target silencing. This could have occurred if there was sufficient sequence similarity between the KS domains of pks1 and swnK to initiate silencing of swnK. While pks1 is a highly conserved nonreducing PKS, swnK is a hybrid PKS-NRPS (Cook et al. 2017). A Blastn comparison of the sequences gave four regions of homology each 17–33 bases with 77–88% identity and a single sequence of 17 bases with 100% identity. Whether that level of similarity is enough to account for the decrease in swainsonine production is not known.
In summary, applicability of using RNAi as a tool for studying gene function in S. leguminicola was demonstrated by the introduction of the IRT pks1 silencing vector into genomic DNA of S. leguminicola cells which resulted in S. leguminicola phenotypes of a reduced melanin production, indicating that pks1 has a functional role in the melanin formation in S. leguminicola.
References
Ahlquist P (2002) RNA-dependent RNA polymerases, viruses, and RNA silencing. Science 296:1270–1273
Akamatsu HO, Chilvers MI, Stewart JE, Peever TL (2010) Identification and function of a polyketide synthase gene responsible for 1,8-dihydroxynaphthalene-melanin pigment biosynthesis in Ascochyta rabiei. Curr Genet 56:349–360
Akihiro M, Makoto U, Sakae A, Junichi K (2007) RNA-mediated gene silencing in the phytopathogenic fungus Bipolaris oryzae. FEMS Microbiol Lett 269:85–89
Alhawatema SM, Sanogo S, Baucom DL, Creamer R (2015) A search for the phylogenetic relationship of the ascomycete Rhizoctonia leguminicola using genetic analysis. Mycopathologia 179:381–389
Aust SD (1974) Rhizoctonia leguminicola-slaframine. In: Purchase IFH (ed) Mycotoxins. Elsevier, Amsterdam, pp 97–109
Aust SD, Broquist HP, Rinehart KL Jr (1966) Slaframine. Structural studies of a parasympathomimetic alkaloid of fungal origin. J Am Chem Soc 88:2879–2880
Bdel-Hadi AM, Caley DP, Carter DRF, Magan N (2011) Control of aflatoxin production of Aspergillus flavus and Aspergillus parasiticus using RNA silencing technology by targeting aflD (nor-1) gene. Toxins 3:647–659
Bernstein E, Caudy A, Hammond S, Hannon G (2001) Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409:363–366
Braun K, Romero J, Liddell C, Creamer R (2003) Production of swainsonine by fungal endophytes of locoweed. Mycol Res 107:980–988
Cook D, Donzelli BGG, Creamer R, Baucom DL, Gardner DR, Pan J, Moore N, Krasnoff SB, Jaromczyk JW, Schardl CL (2017) Swainsonine biosynthesis genes in diverse symbiotic and pathogenic fungi. G3 (Bethesda) 7:1791–1797
Croom WJ, Hagler WM, Froetschel MA (1995) The involvement of slaframine and swainsonine in slobbers syndrome: a review. Anim Sci 73:1499–1508
Crump MH, Smalley EB, Henning JN, Nichols RE (1963) Mycotoxicosis in animals fed legume hay infested with Rhizoctonia leguminicola. J Am Vet Med Assoc 143:996–997
Gardner DR, Cook D (2010) A comparison of alternative sample preparation procedures for the analysis of swainsonine using LC-MS/MS. Phytochem Anal 2:124–127
Gough FJ, Elliott ES (1956) Blackpatch of red clover and other legumes caused by Rhizoctonia leguminicola. W Va Agric Exp Stn Bull 387:1–23
Guengerich FP, Broquist HP (1973) Biosynthesis of slaframine, (1S, 6S, 8aS)-1-acetoxy-6-aminooctahydroindolizine, a parasympathomimetic alkaloid of fungal origin. II. The origin of pipecolic acid. Biochemistry 12:4270–4274
Guengerich FP, Snyder JJ, Broquist HP (1973) Biosynthesis of slaframine, (1S, 6S, 8aS)-1-acetoxy-6-aminooctahydroindolizine, a parasympathomimetic alkaloid of fungal origin. I. Pipecolic acid and slaframine biogenesis. BioChemistry 12:4264–4269
Hagler WM, Croom WJ (1989) Slaframine: occurrence, chemistry, and physiological activity. In: Cheeke PR (ed) Toxicants of plant origin, Volume I, Alkaloids. CRC Press, Boca Raton, FL, pp 257–279
Kawamura C, Moriwaki J, Kimura N, Fujita Y, Fuji S, Hirano T, Koizumi S, Tsuge T (1997) The melanin biosynthesis genes of Alternaria alternata can restore pathogenicity of the melanin-deficient mutants of Magnaporthe grisea. Mol Plant Microbe Interact 10:446–453
Li QQ, Skinner J, Bennett JE (2012) Evaluation of reference genes for real-time quantitative PCR studies in Candida glabrata following azole treatment. BMC Mol Biol 13:22
Liu H, Cottrell TR, Pierini LM, Goldman WE, Doering T (2002) RNA interference in the pathogenic fungus Cryptococcus neoformans. Genetics 160:463–470
Mouyna I, Henry C, Doering TL, Latge JP (2004) Gene silencing with RNA interference in the human pathogenic fungus Aspergillus fumigatus. FEMS Microbiol Lett 237:317–324
Nakayashiki H, Hanada S, Nguyen BQ, Kadotani N, Tosa Y, Mayama S (2005) RNA silencing as a tool for exploring gene function in ascomycete fungi. Fungal Genet Biol 42:275–283
O’Neill NR, Brooker N, Lydon J, Sauders JA (1998) Transformation of Colletotrichum trifolii protoplasts by electroporation and PEG mediated DNA uptake. Recent Res Dev Plant Pathol 2:1–15
Salame TM, Ziv C, Hader O, Yarden Y (2011) RNAi as a potential tool for biotechnological applications in fungi. Appl Microbiol Biotechnol 89:501–512
Smalley EB, Nichols RE, Crump MH. Henning JN (1962) A physiological disturbance in animals resulting from ingestion of Rhizoctonia leguminicola infested red clover forage. Phytopathology 52:753
Wickwire BM, Harris CM, Harris TM, Broquist HP (1990) Pipecolic acid biosynthesis in Rhizoctonia lequminicola. Biol Chem 265:14748 – 1475
Zamore P, Tuschl T, Sharp P, Bartel D (2000) RNAi: double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell 101:25–33
Acknowledgements
This work was financially supported by the New Mexico State University Agricultural Experiment Station and the Molecular Biology Graduate Program. The authors thank the Fungal Genetics Stock Center for kindly providing the fungal silencing vector pSilent-1.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Alhawatema, M.S., Gebril, S., Cook, D. et al. RNAi-mediated down-regulation of a melanin polyketide synthase (pks1) gene in the fungus Slafractonia leguminicola . World J Microbiol Biotechnol 33, 179 (2017). https://doi.org/10.1007/s11274-017-2346-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11274-017-2346-y