
Abstract
Genetic studies are important to understand the complex biological system of various organisms. Some eukaryotic green organisms have tough cell wall which precludes the efficient extraction of the genetic materials. Here, we developed the method for simple and rapid isolation of high quality DNA from a green alga Tetraspora sp. CU2551. The cell homogenization procedures were combined with physical force plus heat treatment to disrupt the cell envelope of Tetraspora sp. CU2551. Without protease treatment, vortexing with glass bead for 30–105 s at 70 °C led to the isolation of a high purity DNA which was suitable for downstream process. The improved method was successfully developed and could be applied for the rapid isolation of DNA from other unicellular and filamentous green microalgal strains.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Tetraspora sp. CU2551 is a unicellular freshwater green alga isolated from natural source of Thailand. This green microorganism was previously characterized showing efficient biohydrogen production through the catalysis of hydrogenase enzyme (Maneeruttanarungroj et al. 2012). The hydrogen gas product could be an invaluable source of alternative energy. Recently, many physical parameters were optimized to maximize the capacity of the production (Maneeruttanarungroj et al. 2010). Not only that the hydrogen gas could be produced by this green microorganism, but our previous study also showed that Tetraspora sp. CU2551 could accumulate the photoprotective compounds which can be used to produce invaluable compounds as the commercial product (Rastogi and Incharoensakdi 2013). Although the production capacity was maximized and the invaluable photoprotective compounds were analyzed, the threshold of those production capacities could also be elevated by genetic manipulation.
DNA-based molecular biology will help to elucidate and enable researchers to fully understand the system in vivo in more detail. Many standard protocols are available for DNA extraction, with different degree of success, such as organic extraction (Sambrook and Russell 2001), sodium dodecyl sulfate (SDS) method (Marmur 1961), cetyltrimethylammonium bromide (CTAB) method (Doyle and Doyle 1990), high salt low pH method (Guillemaut and Maréchal-Drouard 1992), salt extraction method (Aljanabi and Martinez 1997), and NaOH method (Hill-Ambroz et al. 1992). However, some of these standard protocols may be suitable for some specific organisms, for instance, CTAB is suitable for DNA isolation from plants whereas SDS is preferred for DNA isolation from bacteria.
In several algal strains, many chemical-based methods are used in cells lysis for DNA isolation procedures. Many brown algal strains are used in comparison for DNA isolation using CTAB method (Phillips et al. 2001). The marine macroalgae are also extracted for DNA using SDS method (Su and Gibor 1988). The DNA of model organism Chlamydomonas reinhardtii is also isolated by SDS method (Newman et al. 1990). However, those methods used the chemical-based techniques to lyse the membrane system of algae which is not suitable for the specific tough cell wall green alga Tetraspora sp. CU2551.
Our previous observation showed that Tetraspora sp. is unique in that it has a transparent sheath with a tough cell wall (Maneeruttanarungroj et al. 2010). Several methods were applied to disrupt the cell envelope of this alga such as repeating freeze–thaw cycle and cell grinding in mortar. However, the tested methods were inefficient to extract the genetic materials from the cells. The difficulty of the DNA isolation from plants and algae has been reported (Doyle and Doyle 1987). Moreover, a suitable protocol to isolate DNA from one strain often fails with other strains. It is probably because of the diversity of cell wall composition and the presence of various secondary metabolites in those cells (Doyle and Doyle 1990). One method of destroying the membrane organization is the use of glass bead vortexing by the high-cost instrument, Precellys Bead Beater (Bertin technologies, France) with the speed of 5500 rpm for 15 s (Maneeruttanarungroj et al. 2012). Such a high speed of the vortexing enabled the release of high yield of DNA, but the shear force could lower the quality of DNA making this method unsuitable for DNA extraction for Tetraspora sp. In order to obtain the high quality genetic materials with low cost technique, this study aimed to develop the simple and rapid method for isolation of high quality DNA from a green alga Tetraspora sp. CU2551 containing a tough cell wall.
Materials and methods
Cell culture
Tetraspora sp. CU2551 was cultivated in TAP medium under the optimal condition for 24 h as described previously (Maneeruttanarungroj et al. 2010). Briefly, cells from 25 ml culture were harvested by centrifugation at 5000×g for 1 min. The pellet was further used in DNA isolation experiments.
DNA extraction
Three different traditional treatments namely SDS, glass bead, and phenol were used for screening the suitable method in lysing the cells to obtain high yield of DNA. Prior to these treatments, 10 min incubation of 500 µl cell suspension in Tris–EDTA buffer (TE buffer) with or without addition of 1 µl of 50 mg/ml cellulase (ONOZUKA R-10, PhytoTechnology Laboratory® company, USA) was also carried out to test for the effect of cell wall digestion. After incubation with or without cellulase, cells were harvested for DNA isolation. The purified DNA was further monitored for its quality by 1 % agarose gel electrophoresis.
DNA isolation using SDS
Cell pellet was resuspended in 500 µl TE buffer followed by 10 µl of 20 % SDS solution. The cell suspension was incubated at room temperature or at 60 °C for 5 min. Cells were immediately subjected to vortex at highest speed of bench-top vortex mixer for 5 min. An equal volume of phenol solution (phenol: chloroform: isoamyl alcohol at 25:24:1 v/v) was then added to cell suspension and briefly mixed by vortexing followed by centrifugation at 12,000×g for 5 min at room temperature. The supernatant was transferred to a new tube and an equal volume of chloroform was added and briefly mixed prior to centrifugation at the same speed for 5 min. Aqueous phase was transferred to a new tube with addition of an equal volume of isopropanol. The mixture was kept at room temperature for 1 min before centrifugation at 12,000×g for 5 min at 4 °C to collect DNA pellet. The pellet was washed once with 1 ml of ice-cold 75 % ethanol, air dried and dissolved in TE buffer.
DNA isolation using glass bead (GB)
Cell pellet was resuspended in 500 µl TE buffer together with 100 mg of 0.2–0.3 µm glass bead. The cell suspension was incubated at 60 or 95 °C for 5 min before subjecting to vortex at highest speed of bench-top vortex mixer for 5 min. The remaining procedures were as described in section “DNA isolation using SDS”.
DNA isolation using glass bead and phenol solution (PS)
Cell pellet was resuspended in 500 µl of phenol solution (phenol: chloroform: isoamyl alcohol at 25: 24: 1 v/v) together with 100 mg of 0.2–0.3 µm glass bead. The cell suspension was incubated at 60 or 95 °C for 5 min. The tube was immediately subjected to vortex at highest speed of bench-top vortex mixer for 5 min. The remaining procedures were as described in section “DNA isolation using SDS”.
Spectrophotometry analysis
All DNA samples isolated according to section “DNA isolation using SDS” were dissolved in TE buffer. One microliter of sample was added with 99 µl of TE buffer. The diluted samples were analyzed by spectrophotometer (BioPhotometer UV/Vis Spectrophotometer, Eppendorf) at 230, 260 and 280 nm using TE buffer as a blank solution.
PCR analysis
Three gene specific primer pairs for 18S rDNA, sulP and hydA genes were used to monitor the downstream process after DNA isolation. The primer sequences were described elsewhere (Maneeruttanarungroj et al. 2012). PCR cycles were conducted in 20 µl reaction mixtures containing 1× Taq DNA Polymerase buffer (Invitrogen), 8 pmol of each forward and reverse primer, 4 nmol of dNTP, 1 µl of DNA template and 0.4 U of Taq DNA Polymerase (Invitrogen). The PCR program consisted of 30 s at 94 °C followed by 30 cycles of 94 °C for 15 s, 55 °C for 15 s, 72 °C for 30 s, and then a final extension at 72 °C for 1 min. The PCR reaction mixture was electrophoresed in 1 % agarose gel with 0.5× TAE buffer. The DNA band was visualized under UV light by ethidium bromide staining.
DNA extraction by the developed protocol
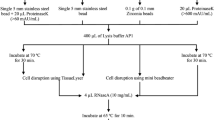
After optimization to obtain a suitable procedure for DNA extraction of Tetraspora sp., the developed protocol, as shown in Fig. 6, was used for DNA extraction from other nine microalgal strains obtained from Thailand Institute of Scientific and Technological Research (TISTR) and Laboratory of Cyanobacterial Biotechnology, Chulalongkorn University, Thailand. The 1–2 weeks old cells were harvested and resuspended in 400 µl TE buffer with 100 mg glass bead prior to incubation at 70 °C for 5 min. The suspension was vortexed at the maximum speed for 30 s followed by the addition of 500 µl phenol solution (phenol: chloroform: isoamyl alcohol at 25: 24: 1 v/v) to eliminate proteins. The mixture was centrifuged to separate the phenol solution. The upper aqueous phase was washed with an equal volume of chloroform. The DNA in aqueous phase was precipitated by 75 % ice-cold ethanol followed by air dry.
Results
Morphology observation
To confirm that the Tetraspora sp. CU2551 has tough cell envelopment, the Tetraspora sp. cell morphology was observed using 400× magnifications under light microscope (Fig. 1). A single whole cell and multiple-cells barrier could be observed with diameter in the range of 5–10 µm. Cell compartmentalizations could be clearly seen confirming that Tetraspora sp. is the eukaryotic organism. The algal cells were encapsulated by the transparent sheath as indicated by arrows. It is believed that this encapsulation system makes the serious problem in DNA extraction from this organism (Maneeruttanarungroj et al. 2010).

Cell morphology observed under light microscope at ×400 magnification. Arrows indicate the transparent sheath encapsulating the Tetraspora sp. CU2551 cells
DNA extraction screening
To obtain the DNA from the alga, a combination of conventional methods was used to screen for an alternative means of suitable isolation. The physical stress, temperature stress and basic chemistry were combined as shown in Table 1. The cellulase-treatment experiment was used for comparative purpose. There are no available data for the genome size of Tetraspora sp. However, previous studies in other green algae have reported the genome size of Chlamydomonas reinhardtii with the size of about 120.4 Mb (Merchant et al. 2007) and of Ostreococcus tauri with the size of about 12.6 Mb (Derelle et al. 2006). Presumably the expected genome size of Tetraspora sp. could be within the range of 12–120 Mb. In Fig. 2, the maximum size of DNA marker is 12 kb, thus the expected band of extracted DNA should be above the marker size. The overall results suggested that cellulase treatment did not improve DNA isolation as compared to the non treatment experiment (Fig. 2). Thus, the enzyme treatment for cell wall digestion was not further explored. Cell lysis by 0.5 % (w/v) sodium dodecyl sulfate (SDS) at both room temperature and 60 °C showed the same efficiency of DNA isolation as depicted in lanes 1 and 2, respectively. Cell incubation in TE buffer at 60 °C for 5 min followed by glass bead vortexing (lane 3) showed the sharp band with the size above 12 kb. This method gave a better yield of DNA than that by phenol solution with glass bead method (lane 5). It should be noted that incubation at 95 °C by both methods caused DNA damage resulting in small fragments as seen by the smear pattern as shown in lanes 4 and 6. These results suggested that optimal vortexing with glass beads combined with incubation at suitable temperature is important for increasing the yield and quality of isolated DNA.

Agarose gel electrophoresis of DNA isolation method. Lane 1–6 DNA from cells without cellulase treatment; lane 7–12 DNA from cells with cellulase treatment. Each lane is described as shown in Table 1. M: 1 kb Plus DNA ladder (Invitrogen)
Optimization
Though the glass bead vortexing after incubating cells at 60 °C for 5 min showed the good quality of DNA with no smear pattern of fragile DNA strand, the optimal temperature and beating time were further investigated to enhance the yield. Figure 3 shows the yield and quality of DNA isolated from Tetraspora sp. under different conditions. Vortexing time of 30 s yielded the cellular DNA with the range of 0.15–0.25 µg/(ml culture) after 5 min incubation at various temperatures ranging from 30 to 80 °C (Fig. 3a). The absorbance ratio of 260/280 and 260/230 were used to monitor the contamination of protein and phenol, respectively. The maximum yield was obtained when incubating cells at 50 °C with the DNA content of about 0.3 µg/(ml culture); however, a small portion of protein and phenol could still be detected. Interestingly, when increasing the incubation temperature to 70 °C, protein contamination was almost negligible as evidenced by an increase of the 260/280 value to about 2.0; however, only one-third DNA yield was recovered compared to that at 50 °C incubation. The good DNA quality was confirmed by agarose gel electrophoresis shown at the top panel of the figure.

The yield (filled circle), protein (filled triangle) and phenol (filled square) contamination of isolated DNA under different conditions. a Effect of temperature at 5 min incubation, b effect of beating time after 5 min incubation at 50 °C and c effect of beating time after 5 min incubation at 70 °C. The top panel of each graph is agarose gel electrophoresis that showed the corresponding DNA obtained from each condition
Moreover, we further tested the effect of the beating time on DNA isolation ranging from 0 to 105 s after incubating cells at both 50 °C (Fig. 3b) and 70 °C (Fig. 3c) for 5 min. For 50 °C incubation (Fig. 3b), increasing beating time up to 105 s resulted in an increase of the DNA yield, especially the yield was drastically increased after 75 s. An increase in the purity of DNA was also obtained with increasing beating time as evidenced by an increase of 260/280 ratio. A high portion of protein contamination was observed without the action of glass beads, i.e. with no beating time at 0 s. Over the range of beating time up to 105 s, no apparent damage on the quality of DNA strand was detected in the gel pattern. Interestingly, when incubating cells at 70 °C, protein contamination was greatly reduced as observed by a high 260/280 ratio and glass beads beating had no effect on the reduction of protein contamination (Fig. 3c). The linear relationship between the beating time and DNA yield could also be observed. As a consequence, purified DNA could be obtained with no protease treatment at 70 °C incubation. The overall results indicated that DNA extraction procedure at 70 °C gave a higher quality of DNA than that at 50 °C, albeit the higher DNA yield obtained at 50 °C.
Characterization
The purified DNA obtained after incubation at both 50 and 70 °C was further investigated for the downstream process by PCR reaction using primers specific for the three genes, 18S rDNA, sulfate permease and hydrogenase A. The PCR product was electrophoresed and showed the same pattern for both 50 and 70 °C incubations with corresponding PCR product size. The PCR pattern of DNA obtained from 70 °C incubation is only shown in Fig. 4. The expected band was observed with the corresponding size of each primer pair. 18S rDNA gene was successfully amplified with all ranges of DNA concentrations tested, but sulP and hydA were successfully amplified with 1 ng and 0.1 ng DNA, respectively. This indicates the inequality of gene copy number distributed in the genome. However, the overall results showed that isolated DNA from this study can be further used in the downstream process.

Agarose gel electrophoresis showing the downstream process after isolation. Various amounts of DNA ranging from 0.1 pg to 1 ng was used as a template in amplification with three gene specific primers. M: GeneRuler™ 100 bp DNA Ladder (Fermentas), 18 s: 18 s rDNA, sulP: sulfate permease gene, hydA: hydrogenase gene
Application to other green microalgal strains
The overall results demonstrated that the suitable condition to destroy the membrane organization of Tetraspora sp. was the use of pretreatment at temperature of 70 °C for 5 min followed by glass bead vortexing for 30–105 s. Our developed protocol is not only suitable for a tough cell wall Tetraspora sp. CU2551, but also could be applied to other microalgal strains. The nine strains used were six filamentous strains and three unicellular strains. The nine strains used according to the lane number of Fig. 5 were Synechocystis sp. PCC 6803, Synechococcus elongatus PCC 7942, Anabaena siamensis TISTR 8012, Anabaena sp. TISTR 8076, Aphanocapsa biformis TISTR 8543, Fischerella muscicola TISTR 8215, Fischerella sp. TISTR 8218, Mastigocladus sp. TISTR 8240 and Oscillatoria sp. TISTR 8657. The results showed that our developed protocol is not only suitable for the tough-cell wall Tetraspora sp. CU2551 (lane T) which has the unicellular morphology, but also for other two unicellular cell morphology strains (lanes 1 and 2) and three filamentous cell morphology strains (lanes 3, 6 and 7). However, our developed protocol failed to extract DNA from four cyanobacteria, Anabaena sp. TISTR 8076, Aphanocapsa biformis TISTR 8543, Mastigocladus sp. TISTR 8240 and Oscillatoria sp. TISTR 8657 (lanes 4, 5, 8 and 9, respectively).

Agarose gel electrophoresis of DNA extracted by the developed protocol from other nine microalgal strains. M: 1 kb Plus DNA ladder (Invitrogen), T: Tetraspora sp. DNA, 1: Synechocystis sp. PCC 6803, 2: Synechococcus elongatus PCC 7942, 3: Anabaena siamensis TISTR 8012, 4: Anabaena sp. TISTR 8076, 5: Aphanocapsa biformis TISTR 8543, 6: Fischerella muscicola TISTR 8215, 7: Fischerella sp. TISTR 8218, 8: Mastigocladus sp TISTR 8240 and 9: Oscillatoria sp. TISTR 8657
Discussion
Since DNA isolation step is a time-consuming process, there have been studies to reduce the time and cost of isolation (Pinto et al. 2009; Pedersen et al. 2012). Tetraspora sp. CU2551 cells are hard to break and to overcome this problem some traditional methods were chosen as method of choices to disrupt the cells such as freeze–thaw cycle repeating or liquid nitrogen grinding. The use of freeze–thaw cycle repeating did not improve the yield of extraction, but liquid nitrogen grinding showed the good yield of the DNA. However, the quality of the obtained DNA was not reproducible since the physical force in grinding could shear the DNA molecules. The sheared DNA can be monitored on agarose gel electrophoresis (Yuan et al. 2012). The small degraded DNA could be observed down the bottom of agarose gel when grinding cells for longer time (data not shown).
Tetraspora sp. CU2551 is a suitable organism for in-depth study in terms of genetic information since its metabolism can yield some valuable secondary metabolites such as hydrogen gas which can be further used as an alternative energy source (Maneeruttanarungroj et al. 2010). The study started at the screening step to find the suitable method for cell membrane disruption. In the procedure, ONOZUKA R-10 cellulase was used in cell pre-treatment to digest cell wall. On the other hand, this cellulase contains a combination of cellulase, hemicellulase, pectinase, α-amylase, and protease enzymes which can lead to digestion of not only cellulose on the cell wall, but also cellular proteins. Due to the mixing of several enzymes, this cellulase was widely used in cell wall digestion (Bondino and Valle 2009; de Castro et al. 2011; Konwarh et al. 2012). However, cellulase had no significant effect on the yield of DNA extraction of Tetraspora sp. Not only cellulase had no significant impact in extraction procedure but sodium dodecyl sulfate (SDS) which is a widely used anionic surfactant to lyse the cell membrane also showed no significant efficiency of the extraction. This is because the transparent sheath of the cell envelope may block the chemical penetration to cell membrane. In contrast, phenol solution could destroy the cell organization. This is due to the fact that the organic solvent can denature most of biomolecules such as protein (Macdonald and Khajehpour 2015). The phenol solution used in this experiment contains iso-amyl alcohol which can prevent foaming between the organic and aqueous phases which sometimes occurs when only pure phenol–chloroform was used. Although the phenol can denature impurities such as protein molecules, the smear pattern of DNA was observed when incubating at 95 °C. This pattern was also observed when isolating DNA by glass bead vortexing at 95 °C. This was ascribed to the fact that too high temperature can irreversibly damage most biomolecules since the heat can destroy the inter- and intra- molecular interactions such as hydrogen bonding. The overall results suggest that disruption of cell membrane could be achieved by heating up the cells in TE buffer followed by vortexing for several minutes. Heating up to 50–70 °C, which has high potential to disrupt the membrane organization, in combination with vortexing with glass bead to generate physical force can be used as an effective method to release the genetic materials from cells with tough cell wall.
The optimization was performed to reduce time and cost for cell breaking. Here, we propose the DNA extraction procedure as shown in Fig. 6. The suitable temperature and time to obtain high quality of DNA are 70 °C and 5 min, respectively, with further vortexing in the presence of glass beads at the highest speed for 30–105 s. However, in case the high yield of DNA is desired the 50 °C incubation temperature should be used.

The flow chart showing the overall process for DNA isolation procedure developed in this study
After DNA isolation procedure, in general, the polysaccharides, polyphenolic compounds and other secondary metabolites can co-precipitate with the DNA molecule. Those precipitants can inhibit the down-stream enzymatic reactions resulting in the failure use of DNA (Huang et al. 2000). This study showed that the isolated DNA from our developed protocol could be successfully used in the down-stream enzymatic reaction through polymerase chain reaction (PCR). The result also showed the success in amplification reaction using three primers. This is to confirm that our developed protocol can isolate the high quality DNA for the use in other purposes.
In conclusion, the high quality DNA could be obtained from our successfully developed procedure for the tough-cell wall model organism Tetraspora sp. CU2551 with low-cost and short time procedure. Moreover, the isolated genetic materials are suitable for the downstream processes which are normally performed after the extraction. Interestingly, these developed methods could be applied to other green microalgae.
References
Aljanabi SM, Martinez I (1997) Universal and rapid salt-extraction of high quality genomic DNA for PCR-based techniques. Nucleic Acids Res 25:4692–4693
Bondino HG, Valle EM (2009) A small intergenic region drives exclusive tissue-specific expression of the adjacent genes in Arabidopsis thaliana. BMC Mol Biol 10:95
de Castro LM, Mourao FDA, Mendes BMJ (2011) Isolation and protoplasts plating efficiency of sweet orange. Rev Bras Fruticult 33:509–516
Derelle E et al (2006) Genome analysis of the smallest free-living eukaryote Ostreococcus tauri unveils many unique features. Proc Natl Acad Sci USA 103:11647–11652
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19:11–15
Doyle JJ, Doyle JL (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Guillemaut P, Maréchal-Drouard L (1992) Isolation of plant DNA: a fast, inexpensive, and reliable method. Plant Mol Biol Rep 10:60–65
Hill-Ambroz KL, Brown-Guedira GL, Fellers JP (1992) Modified rapid DNA extraction protocol for high throughput microsatellite analysis in wheat. Crop Sci 42:2088–2091
Huang JC, Ge XJ, Sun M (2000) Modified CTAB protocol using a silica matrix for isolation of plant genomic DNA. Biotechniques 28:432–434
Konwarh R, Pramanik S, Kalita D, Mahanta CL, Karak N (2012) Ultrasonication—a complementary ‘green chemistry’ tool to biocatalysis: a laboratory-scale study of lycopene extraction. Ultrason Sonochem 19:292–299
Macdonald RD, Khajehpour M (2015) Effects of the protein denaturant guanidinium chloride on aqueous hydrophobic contact-pair interactions. Biophys Chem 196:25–32
Maneeruttanarungroj C, Lindblad P, Incharoensakdi A (2010) A newly isolated green alga, Tetraspora sp. CU2551, from Thailand with efficient hydrogen production. Int J Hydrogen Energy 35:13193–13199
Maneeruttanarungroj C, Lindblad P, Incharoensakdi A (2012) Sulfate permease (SulP) and hydrogenase (HydA) in the green alga Tetraspora sp CU2551: dependence of gene expression on sulfur status in the medium. Int J Hydrogen Energy 37:15105–15116
Marmur J (1961) A procedure for the isolation of deoxyribonucleic acid from micro-organism. J Mol Biol 3:208–218
Merchant SS et al (2007) The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science 318:245–250
Newman SM, Boynton JE, Gillham NW, Randolph-Anderson BL, Johnson AM, Harris EH (1990) Transformation of chloroplast ribosomal RNA genes in Chlamydomonas: molecular and genetic characterization of integration events. Genetics 126:875–888
Pedersen IS, Krarup HB, Thorlacius-Ussing O, Madsen PH (2012) High recovery of cell-free methylated DNA based on a rapid bisulfite-treatment protocol. BMC Mol Biol 13:12
Phillips N, Smith CM, Morden CW (2001) An effective DNA extraction protocol for brown algae. Phycol Res 49:97–102
Pinto FL, Thapper A, Sontheim W, Lindblad P (2009) Analysis of current and alternative phenol based RNA extraction methodologies for cyanobacteria. BMC Mol Biol 10:79
Rastogi RP, Incharoensakdi A (2013) UV radiation-induced accumulation of photoprotective compounds in the green alga Tetraspora sp. CU2551. Plant Physiol Biochem 70:7–13
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Su X, Gibor A (1988) A method for RNA isolation from marine macro-algae. Anal Biochem 174:650–657
Yuan SQ, Cohen DB, Ravel J, Abdo Z, Forney LJ (2012) Evaluation of methods for the extraction and purification of DNA from the human microbiome. PLoS One 7(3): e33865
Acknowledgments
This work was supported by King Mongkut’s Institute Technology Ladkrabang fund (KREF015701) to C. Maneeruttanarungroj, the new researcher fund. The research grants to A. Incharoensakdi provided by Chulalongkorn University Ratchadaphiseksomphot Endowment Fund on the Frontier Research Food and Water Cluster (CU-58-011-FW) and by the Thailand Research Fund (IRG 5780008) are also acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Maneeruttanarungroj, C., Incharoensakdi, A. Rapid method for DNA isolation from a tough cell wall green alga Tetraspora sp. CU2551. World J Microbiol Biotechnol 32, 99 (2016). https://doi.org/10.1007/s11274-016-2055-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11274-016-2055-y