
Abstract
African monkeys are resistant to HIV-1 infection due to intrinsic restriction mechanisms found in their cells. However, although they can be infected by monkey-adapted modified HIV-1 particles that are designed to overcome known restriction factors, virus numbers drop to undetectable levels in immunocompetent animals. These results indicate the possibility of the presence of yet unidentified factor(s) that restrict HIV-1 in old-world monkey (OWM) cells after integration of the viral genome into the host cell chromosome. In the light of these findings, we hypothesized that OWMs might have evolved resistance mechanism(s) against HIV-1 by switching specific gene(s) on in response to the synthesis of viral proteins in infected cells. In an attempt to mimic post-infection status, we expressed HIV-1 Tat gene in African green monkey cells and compared the whole proteome with normal cells and identified secretory leukocyte protease inhibitor (SLPI), a protein with known extracellular anti-HIV-1 activity, as an over-expressed protein in the presence of HIV-1 Tat protein by 2D-PAGE and mass spectrometry analysis. We also showed that overexpression of SLPI in the presence of HIV-1 Tat was specific to monkey cells. Our results also suggest that SLPI had a previously undiscovered intracellular anti-HIV activity in addition to its extracellular activity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Human immunodeficiency virus type-1 (HIV-1), the cause of AIDS in humans, is derived from a clade of lentiviruses known as simian immunodeficiency viruses (SIVs), found naturally in species of old-world monkeys (OWMs) in Africa. It is estimated that OWMs were infected by SIVs as far back as 16 million years ago [1]. During this period, OWMs evolved mechanism(s) that protect them from developing SIV related diseases [2]. Thus, while OWMs can be efficiently infected by their species-specific SIVs without any apparent symptoms, HIV-1 infections are pathogenic in humans, since human populations acquired HIV-1 only around 100 years ago [3].
Although OWMs can be infected by their species-specific SIVs, they are resistant to infection by HIV-1. This is perhaps due to exposure of OWMs to past infections by HIV-1-like viruses that led to the emergence of restriction mechanism(s) against HIV-1. HIV-1 can effectively enter the cells of humans and OWMs including African Green Monkey (AGM) and macaques. However, it is inefficient in replicating and inducing pathogenesis in monkeys due to the restriction of the virus in cells by several cellular proteins which operate before the integration of the viral genome into the host chromosome [4,5,6]. Characterization of HIV-1 host restriction factors led to the construction of modified viruses designed to overcome known restriction factors in an attempt to develop animal models of AIDS in macaques. However, although these monkey-adapted viruses initially efficiently replicated in animals, virus numbers dropped to undetectable levels unless the animals were immunocompromised by CD8 cell depletion during and after experimental infections [7, 8]. These results indicate the possibility of the presence of yet unidentified factor(s) that restrict HIV-1 in OWM cells after integration of the viral genome into the host cell chromosome. In the light of these findings, we hypothesized that OWMs might have evolved resistance mechanism(s) against HIV-1 by switching specific gene(s) on or off in response to the synthesis of viral proteins in infected cells. This strategy would not only help the cellular economy but could also protect the cells from the potential side effects of anti-viral genes unless the cells were infected with the virus.
HIV-1 Tat is among the first viral proteins to be synthesized in the infected cells and it regulates HIV-1 gene expression. Specifically, Tat is a transcription activator of the viral LTR promoter, and it is also known to affect the expression of several host genes in the infected cells [9], making it a strong candidate for the activation of potential host resistance genes. To test our hypothesis, we aimed to identify over-expressed proteins in HIV-1 tat-expressing AGM cells by two-dimensional polyacrylamide gel electrophoresis and mass spectrometry and investigate their potential anti-HIV-1 effects. Here we report the overexpression of secretory leukocyte protease inhibitor (SLPI), a protein with known extracellular anti-HIV-1 activity [10,11,12], in AGM but not in human cells in the presence of HIV-1 Tat protein. We also showed that overexpression of SLPI in both human and AGM cells led to lower production of HIV-1 in single-round infection delineating a previously unknown intracellular effect of SLPI.
Material and methods
Plasmids and antibodies
Mammalian expression vector pBud-CE4.1 and luciferase expression plasmid pNL3.2.NF-kappaB-RE were purchased from Thermo Fisher Scientific and Promega, respectively. Plasmids pCV1, pHIVlacZ, which drives the expression of the LacZ gene from HIV-1 LTR promoter and pNL4.3 (a molecular clone of HIV-1) were obtained from the NIH AIDS reagent program (www.aidsreagent.org). pHIV-luc plasmid, which drives the expression of nano luciferase gene from the HIV-1 LTR promoter, was constructed as follows: Nanoluciferase gene, amplified by PCR using primers Luc_F (5′-AAGCTTGCCACCATGGTCTTCACACTC-3′), and Luc_R (5′-GAATTCCCCAATACGCAAACGGATCC-3′) from plasmid pNL3.2.NF-kappaB-RE was restriction digested with HindIII and MunI enzymes and cloned in place of the LacZ gene in pHIVLacZ plasmid digested with enzymes HindIII and EcoRI. HIV-1 Tat expression plasmid pBud-Tat was constructed as follows: PCR amplified Tat cDNA from plasmid pCV1 using primers Tat_F (5′-GCGGCCGCACCATGGAGCCAGTAGATC-3′) and Tat_R (5′-AGATCTATTCCTTCGGGCC-3′) that contained NotI and BglII restriction enzyme sequences (underlined), respectively, was digested with NotI and BglII and cloned downstream the EF-1α promoter in pBud-CE4.1 vector linearized with the same enzymes. The functionality of the cloned HIV-1 Tat protein was confirmed by luciferase assay on the extracts of 293T cells co-transfected with pHIV-luc plasmid and either with empty vector pBud-CE4.1 or Tat-expressing plasmid pBud-Tat (Fig. 1). Luciferase expression in plasmid pHIV-luc is driven by the HIV-1 LTR promoter which is induced by Tat protein. Luciferase activity was about 15 times higher in cells transfected with Tat expression plasmid compared with cells that contained empty vector, confirming the functionality of the cloned HIV-1 Tat.

Luciferase activity of 293T cell extracts transfected with indicated plasmids, relative to pHIV-luc transfected cells. LacZ: pHIV-luc; empty: pBud-CE4.1; Tat: pBud-CE4.1-Tat. (*p < 0.005). The experiments were repeated three times in duplicates, and the data shown are the mean of the data from three experiments
Plasmid pBud-SLPIagm which expresses SLPI gene from AGM was constructed as follows: AGM SLPI (SLPIagm) cDNAs were amplified by reverse transcriptase PCR from total RNA extracts of CV-1 cells using the primer pair SLPI_F: 5′-CTGCAGCACCATGAAGTCCAGYGGCC-3′ and SLPI_R: 5′-TCTAGATCAAGCTTTCACAGGGGAAACG-3′, which contained restriction enzyme sites PstI and XbaI (underlined), respectively. Restriction enzyme digested cDNAs were then cloned downstream the CMV promoter in pBud-CE4.1 vector linearized by the same restriction enzymes to generate plasmid pBud-SLPIagm.
Rabbit polyclonal antibodies against human SLPI (also reacts with mouse SLPI) (PA5-20385), human GAPDH (PA1-987) and HRP-linked anti-rabbit/mouse IgG (31464) were purchased from Thermo Fisher Scientific. Rabbit polyclonal antibodies against HIV-1 Tat (705) and GST-Tat fusion protein (2367) were obtained from NIH AIDS Reagent Program.
Cell culture and transfection
CV-1 (Chlorosebus tantalus kidney) [13, 14], COS7 (a derivative of CV-1 that expresses SV40 large T antigen), Vero (Chlorocebus sabaeus kidney) [15], HeLa (human epithelial cervical carcinoma) and 293T (human embryonic kidney cell line that expresses both adenovirus E1A/E1B and SV40 T antigen) [16] cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS) and 2 mM l-glutamic acid. All cells were incubated in 5% CO2 and 90–100% relative humidity at 37 °C. Confluent cultures were passaged at a ratio of 1:4 or 1:6 every 2–3 days with the use of trypsin–EDTA (0.25% and 0.02%, respectively). High-quality plasmid DNAs suitable for transfection were purified using PureLink HiPure plasmid midiprep kit (Thermo Fisher Scientific). One day after seeding of 2 × 105 cells into 24-well plates, all cells were transfected with 250 ng of each plasmid using TurboFect™ in vitro transfection reagent (Thermo Fisher Scientific). Transfected cells were analyzed 48 h post-transfection. Transfection efficiencies of cell lines were determined and shown to be similar by qRT-PCR analysis with specific primers for the zeocin resistance gene present in the plasmids (data not shown).
Luciferase activity assay
Luciferase reporter assay was performed in an ARVO X plate reader (PerkinElmer), using Nano-Glo® Luciferase Assay System, according to the manufacturer’s instructions (Promega). Data were measured as the ratio of luminescence from the experimental reporter to the luminescence from the control reporter and normalized to control wells.
Protein extraction from cultured cells
Proteins were extracted from cultured cells lysed with ProteoJET™ Mammalian Cell Lysis Reagent containing Halt™ protease inhibitor cocktail (Thermo Fisher Scientific), by cell centrifugation at 12,000×g at 4 °C for 10 min. The supernatants were collected as the whole cell extracts and stored at − 80 °C for further use. Protein concentration was determined by using the Bradford protein assay kit (Thermo Fisher Scientific).
Separation of proteins by two-dimensional polyacrylamide gel electrophoresis (2D-PAGE)
Two hundred and fifty micrograms of protein extracts from cultured cells were loaded onto IPG strip (Bio-Rad Laboratories) and subjected to isoelectric focusing using PROTEAN IEF CELL (Bio-Rad Laboratories) at 20 ℃. The IEF program was as follows: 50 V, 2 h; 300 V, 2 h; 500 V, 1 h; 1000 V, 2 h and then 8000 V for 1 h with the maximum current limit of 50 µA. Following IEF, strips were equilibrated two times for 20 min, first in 20 ml of 50 mM Tris–HCl (pH 8.8), containing 6 M urea, 20% glycerol, 2% SDS (sodium dodecylsulfate) and 2% w/v DTT (dithiothreitol), and then in same buffer containing 2.5% w/v IAA (iodoacetamide) instead of DTT. Strips were then subjected to second dimension gel electrophoresis on a 12% SDS-PAGE gel (1 mm gel thickness) using a vertical electrophoresis unit (Protean II xi 2-D Cell, Bio-Rad Laboratories) at a constant voltage of 90 V until the dye front reached 0.5 cm above the bottom of the gel. The gels were fixed in a mixture of 40% v/v ethanol and 10% v/v glacial acetic acid for 12 h with continuous shaking and then the proteins were visualized by Coomassie blue staining. Differentially expressed protein spots were determined visually and excised from the gel for mass spectrometry analysis.
Protein identification by mass spectrometry
Protein spots excised from the 2D-PAGE gel of CV1 cell proteome in the pI range of 3–10 were taken separately into a microcentrifuge tube and rinsed with wash solution (50% methanol, 5% acetic acid) in room temperature by several changes of the solution until the dye was removed from the gel. Following dehydration of the gels with acetonitrile and drying under vacuum, proteins in the gel spots were first reduced with a solution containing 100 mM ammonium bicarbonate and 10 mM DTT and then alkylated with a solution containing 100 mM iodoacetamide and 100 mM ammonium bicarbonate. In-gel digestion of the proteins was carried out overnight at 37 °C using 20 µg/ml trypsin solution prepared in 50 mM ammonium bicarbonate. Then, peptides were extracted from each gel piece separately and desalted with C18-ZipTip (Millipore) before analysis. Mass spectrometry analysis of the peptides was carried out with the Autoflex III smart beam MALDI TOF/TOF MS (Bruker Daltonics, Germany) instrument. Two-layer matrix preparation method was used and alpha-cyano-4-hydroxycinnamic acid (CHCA) was prepared as a matrix solution. The mass spectra were recorded in reflectron positive ion mode and each peptide peak was selected for MS/MS analysis using argon as the collision gas. The data obtained from MS/MS analysis were searched against the SwissProt database (version 2015_06) (https://www.uniprot.org/) using probability-based search engine Mascot (version 2.5) (https://www.matrixscience.com) for identification of the corresponding proteins.
Total RNA extraction and real-time quantitative reverse transcriptase PCR (qRT-PCR)
Total RNAs from cells were isolated by using GeneJet RNA Purification Kit (Thermo Fisher Scientific), and residual traces of genomic DNAs were removed from RNA samples using RNase free DNase I (Thermo Fisher Scientific) according to manufacturer’s protocol. Complementary DNAs (cDNAs) were synthesized from 500 ng of total RNA by using the RevertAid™ H Minus First Strand cDNA Synthesis Kit (Thermo Fisher Scientific) with oligo(dT) primer according to manufacturer’s protocol. qRT-PCR was carried out in Lightcycler 480 instrument (Roche Life Sciences) with gene-specific primers using Maxima™ SYBR Green qPCR Master Mix (Thermo Fisher Scientific). The PCR condition was as follows: 1 cycle at 50 °C for 2 min, 1 cycle at 95 °C for 10 min, 40 cycles at 95 °C for 15 s, 62 °C for 30 s, and 72 °C for 30 s. One-tenth of the products were analyzed on 2% agarose gel. mRNA levels of the amplified genes were calculated using the 2(-Delta Delta C(T)) (2T−ΔΔC) method [17]. Primers used for the amplification of target genes were as follows: SLPI, 5′-GCATCAAATGCCTGGATCCT-3′ (forward for both human and monkey), 5′-GCATCAAACATTGGCCATAAGTC-3′ (reverse for human) and 5′-GCATCATACATTGGCCGTAAGCC-3′ (reverse for monkey); GAPDH, 5′-AGAAGGCTGGGGCTCATTTG-3′ (forward for both human and monkey) and 5′-AGGGGCCATCCACAGTCTTC-3′ (reverse for both human and monkey); Zeocin resistance gene, 5′-TGATGAACAGGGTCACGTCGT-3′ (forward) and 5′-AAGTTGACCAGTGCCGTTCCG-3′ (reverse); HIV-1 gag, 5′-AGTRGGGGGACAYCARGCAGCHATGCARAT-3′ (forward) and 5′-TACTAGTAGTTCCTGCTATRTCACTTCC-3′ (reverse) [18]; HIV-1 tat, 5′-AGATCTAGACTAGAGCCCTGGAA-3′ (forward) and 5′-CAAACTTGGCAATGAAAGCAACAC-3′ (reverse).
Genomic DNA purification
Genomic DNA from cells were isolated by using GeneJet genomic DNA Purification Kit (Thermo Fisher Scientific), according to manufacturer’s protocol.
SDS-PAGE and Western blot analysis
Twenty-five micrograms of total proteins extracted from cultured cells were separated on 10% SDS polyacrylamide gel and then transferred onto nitrocellulose membranes (Thermo Fisher Scientific). The membranes were blocked in PBST (phosphate-buffered saline with 0.1% Tween-20) containing 5% nonfat milk for 2 h at room temperature and then probed with primary antibodies. After overnight incubation, the membranes were washed with PBST and further incubated with the corresponding secondary antibodies conjugated with horseradish peroxidase followed by extensive washing with PBST. Immunoreactivity was visualized by using SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific) according to the manufacturer's protocol and the results were recorded by VersaDoc Molecular Imager (Bio-RAD).
Results
Identification of SLPI as a differentially expressed protein in CV-1 cells transfected with HIV-1 Tat expression vector
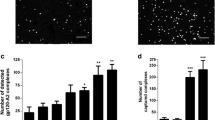
In order to find possible over-expressed host genes in the presence of HIV-1 Tat protein, we performed a comparative analysis of total proteins extracted from AGM CV-1 cells transfected with either tat-expressing or empty vector plasmids after separation by two-dimensional polyacrylamide gel electrophoresis (2D-PAGE). Proteins spots that were distinctly over-expressed only in Tat-expressing cells were isolated from the 2D gel (Fig. 2) and identified by MALDI-TOF–MS–MS, based on peptide mass matching (Table 1). One of the spots numbered 15 in Fig. 2b was identified as the secretory leukocyte protease inhibitor (SLPI) with high MOWSE score by MASCOT. SLPI is a small acidic protein secreted by many cell types including epithelial and dendritic cells [19]. The position of the spot 15 on 2D gel matched the expected isoelectric point (pI) and molecular weight of the SLPI protein, which were 9 and 14 kD, respectively. SLPI caught our attention since it was previously identified as a secreted protein with diverse functions including anti-inflammatory and anti-HIV-1 activities [20]. Therefore, we focused our attention on SLPI as a possible anti-HIV-1 factor that is induced by the presence of HIV-1 Tat protein in AGM cells.

2D-Page analysis and comparison of the total proteome from; a CV-1 cells transfected with the empty expression vector pBud-CE4.1, and b CV-1 cells transfected with the HIV-1 Tat-expressing plasmid pBud-CE4.1-Tat. Proteins overexpressed in CV-1-Tat cells are encircled and numbered. Protein spot 15 was identified as SLPI. c Confirmation of HIV-1 Tat protein production in CV1-Tat cells. Same protein extracts used in a and b were subjected to Western blot analysis using antibodies against Tat. GST-Tat fusion protein was used as positive control
SLPI expression is induced by HIV-1 Tat protein in monkey cells but not human cells
To confirm the induction of SLPI expression by Tat protein, we performed real-time quantitative reverse transcriptase PCR (qRT-PCR) using gene-specific primers on total RNA extracted from pBud-Tat transfected and empty vector transfected monkey and human cell lines. To examine whether the effect of Tat on SLPI expression in AGM cells is unique to the CV-1 cell line, we compared SLPI expression levels of this cell line with two additional cell lines derived from AGMs, (COS-7, a derivative of CV-1 cells and belong to same species, Chlorocebus tantalus and Vero cells, derived from the same tissue (kidney) but from a separate AGM species, Chlorocebus sabaeus) and two human cell lines (HeLa and 293T) (Fig. 3). In each of the AGM cell lines (CV-1, COS7 and vero), SLPI mRNA’s were increased between 10–100 fold in the presence of HIV-1 Tat protein, indicating that the effect of Tat on SLPI expression was conserved in these AGM species, whereas no change was observed in human (HeLa and 293T) cell lines, indicating a species-specific response by AGM cells to HIV-1 Tat protein, hinting to a possible evolutionary adaptation against HIV-1 infection.

HIV-1 Tat protein induces the expression of SLPI in AGM (a) but not human cells (b). Cells, transfected with pBud-Tat (tat) or with empty vector (mock), were cultured for 48 h and subjected to reverse transcriptase qPCR analysis with specific primers for SLPI, and GAPDH. The expressions of SLPI mRNAs in each cell line was normalized to GAPDH expression in the corresponding cells and is shown in graphs as mean ± s.d. Even transformation of cells with the plasmids was confirmed by analyzing the expression of zeocin resistance gene which was present in both the empty and Tat-expressing plasmids. Zeocin resistance gene expression levels in all transfected cells were similar (data not shown). The experiments were repeated three times in duplicates, and the data shown are the mean of the data from three experiments
We further analyzed SLPI expression by western blot, using anti-SLPI polyclonal antibodies on total proteome extracted from pBud-Tat or empty vector transfected AGM and human cell lines (Fig. 4). In each of the AGM cell lines (CV-1, COS7, and vero), SLPI protein levels were considerably increased in the presence of HIV-1 Tat protein, whereas no change was observed in human (HeLa and 293T) cells, validating our qRT-PCR data.

SLPI production is increased in AGM cells in the presence of HIV-1 Tat. a Western blot analysis of SLPI protein in AGM and human cells transfected with either pBud-Tat ( +) or empty vector (−). Total protein was isolated from cells 48 h after transfection and subjected to Western blot analysis using antibodies against SLPI. Membranes were re-probed with anti-GAPDH antibodies to ensure, if any, the effect of HIV-1 Tat expression and even protein loading in each lane. The doubling of the SLPI protein band could be the result of alternative splicing of the SLPI mRNA or post-translational processing of SLPI protein in AGM cells. b Statistical analysis of western blot data (n = 3 of each) was performed using SPSS (Version16.0 Inc, USA). One-way ANOVA was used to analyze the protein levels of SLPI between Tat and empty vector transfected cell lines, followed by Tukey's post-hoc test using the Graph pad prism software Inc., (Version 7.0, California, USA). Western Blot results were expressed as mean ± SEM (standart error of mean). Statistically differences were considered to be significant at *p < 0.05, **p < 0.01 and ***p < 0.001
HIV-1 infection leads to a higher induction of SLPI mRNA expression in CV-1 cells compared to 293T cells
Since Tat protein is produced in HIV-1 infected cells, we investigated the expression of SLPI mRNA in HIV-1 infected CV1 and 293T cells. Both cell types are unsusceptible for HIV-1 infection due to the absence of the CD4 receptor required for HIV-1 binding to susceptible cells [21, 22]. Furthermore, CV1 cells are normally resistant to HIV-1 infection due to premature uncoating of virus particles and prevention of reverse transcription, mediated by the species-specific restriction factor Trim5alpha [23]. To overcome these obstacles, we transfected both CV1 and 293T cells with pNL4-3, a molecular clone of HIV-1, in order to achieve efficient infection [24]. Then, we performed qRT-PCR using SLPI-specific primers on total RNA extracted from pNL4-3 and mock-transfected CV-1 and 293T cells (Fig. 5a). SLPI mRNA expression was induced five-fold in CV-1 cells transfected with pNL4-3 compared to mock-transfected CV-1 cells. However, contrary to our expectations, there was a two and a half-fold increase in SLPI mRNA levels in pNL4-3 transfected 293T cells compared to mock-transfected 293T cells. These results indicate that, unlike in Tat-expressing 293T cells, SLPI expression is induced in HIV-1 infected 293T cells, albeit to a lesser extent compared to HIV-1 infected CV-1 cells.

HIV-1 infection induces SLPI mRNA expression in CV-1 and 293T cells. pNL4-3 (pNL) or mock-transfected cells were cultured for 24 h. Total RNA and DNA were purified from cell lysates and analyzed by RT qPCR for SLPI, Tat and GAPDH from RNA template and by qPCR for HIV-1 Tat and GAPDH from DNA template. a Relative SLPI mRNA levels in mock and pNL4-3 transfected cells. b Confirmation of efficient and even infection of cells by HIV-1 following transfection with pNL4-3 was performed by analyzing DNA levels for HIV-1 tat gene. c Confirmation of HIV-1 Tat expression in pNL4-3 transfected cells. The expression of SLPI and Tat mRNAs, and the amount of tat DNA in each cell line was normalized to GAPDH mRNA and DNA, respectively in the corresponding cells and was shown as mean ± s.d. The experiments were repeated three times in duplicates, and the data shown are the mean of the data from three experiments. Statistically differences were considered to be significant at *p < 0.05, **p < 0.01 and ***p < 0.001
AGM SLPI (SLPIagm) overexpression leads to decreased expression of HIV-1 gag in CV-1 and 293T cells transfected with HIV-1 molecular clone pNL4-3
It was shown that human SLPI protein prevented HIV-1 infection of susceptible human cells by its ability to modulate the interaction of the CD4 receptor with scramblases [11, 12]. The known anti-HIV-1 effect of SLPI is thus extracellular. There is no information regarding any intracellular effects of SLPI on HIV-1 infection and production. Overexpression of SLPI in AGM cells in the presence of HIV-1 Tat, suggests that SLPI may have previously unknown anti-HIV-1 effects in infected cells. We, therefore, examined whether AGM SLPI protein had any effect on HIV-1 production in both human and AGM cells transfected with the molecular clone of HIV-1 (pNL4-3). To evaluate the effect of SLPI protein on HIV-1 production, we transfected the cells with pNL4-3 and compared the expression of the HIV-1 gag gene in SLPIagm over-expressed cells with normal cells. We found that gag-specific mRNAs in SLPIagm overexpressed AGM (CV-1) and human (293T) cells were reduced three- and six-fold, respectively, compared with the normal cells (Fig. 6a). This result showed that SLPIagm was effective on HIV-1 transduced cells of both human and AGM origin in leading to a reduction in gag expression and hence virus production. SLPI and Tat specific mRNA productions were also measured in order to monitor and confirm the expression of the SLPI and Tat in transfected cells (Fig. 6b, c).

The effect of overexpression of SLPIagm protein on HIV-1 gag gene expression in pNL4-3 transfected cells. Cells (CV1 and 293T), transfected with pNL4-3 (pNL) or pNL4-3 and pBud-SLPI (SLPI) or with empty vector, were cultured for 48 h and subjected to reverse transcriptase qPCR analysis with specific primers for GAG, SLPI, Tat and GAPDH. The expressions of GAG, SLPI and Tat mRNAs in each cell line was normalized to GAPDH expression in the corresponding cells and is shown in graphs (a), (b) and (c), respectively as mean ± s.d. SLPI expression (b) was analyzed to confirm the overexpression of SLPI in pBUD-SLPI transfected cells, and Tat specific mRNAs (c) were analyzed to confirm the expression of tat gene in pNL4-3 transduced cells. The experiments were repeated three times in duplicates, and the data shown are the mean of the data from three experiments. Statistically differences were considered to be significant at *p < 0.05, **p < 0.01 and ***p < 0.001
Discussion
HIV-1 variants constructed to overcome restriction factors known to exist in OWMs were able to replicate in monkeys but could not establish persistent infection unless the animals were immunocompromised [7, 8, 25, 26] tipping the immune balance in favor of the virus. This outcome suggests the presence of yet unidentified immune mechanisms in OWMs and prompted us to investigate whether AGM cells produced any intrinsic factor(s) with anti-HIV-1 activity in response to HIV-1 Tat protein that would mimic HIV-1 infection. We chose Tat since it is among the first viral proteins produced in infected cells and has the ability to modulate both viral and cellular gene expression [27].
Our results showed clearly the overexpression of SLPI in different AGM cells expressing HIV-1 Tat protein or infected by an HIV-1 molecular clone. However, there were big differences in the amount of the induction of SLPI mRNA expression in different AGM cell lines (Fig. 3). Induction was 120-fold in CV1 cells, 35-fold in Vero cells and 13-fold in Cos7 cells. Since Cos7 cells are a derivative of the CV1 cells that express SV40 large T antigen it was possible that SLPI induction was somehow affected by the large T antigen. It was shown that SV40 large T expression resulted in the induction of interferon stimulated genes (ISG)s [28], and ISGs are known to cause the inhibition of protein synthesis in the cells [29]. On the other hand, Vero and CV1 cells belong to different species of the AGMs and the differences in SLPI mRNA induction in those cells could simply be due to this fact. The difference of SLPI induction in AGM cell lines was also evident at the protein level and correlated with the mRNA levels (Fig. 4).
Although it is very well established that in human cells, Tat can regulate the expression of many cellular genes by several mechanisms [27], whether the overexpression of SLPI was mediated directly by Tat or indirectly by other Tat-responsive factors remains to be determined. However, the involvement of interferons is unlikely since interferons are generally produced in response to viral structural proteins. Whichever the mechanism, our results suggest that AGMs are evolved to express SLPI in response to HIV-1 Tat protein, at least in the cell types we tested. Tat responsiveness of other AGM cell types, particularly T-cells and macrophages in terms of SLPI expression remains to be determined.
Although there was no change in the expression of SLPI in Tat-expressing human cells we tested, there was a two and a half-fold increase in SLPI in human cells infected with the molecular clone of HIV-1 (Figs. 3 and 5). This increase which was very low compared with the five-fold increase seen in AGM cells could be explained by the findings of Jana et al. [30]. In their study, HIV-1 virus particles incubated with human oral epithelial cells was shown to increase SLPI expression independent of infection as a result of the interaction of the gp120 protein in the virus envelope with the cell surface. The interaction was shown to be completely extracellular and could also be mediated by inactivated virus particles. Therefore, the increase in SLPI expression we observed in human cells was likely due to virus particles produced by HIV-1 transduced human cells but not due to Tat protein.
We showed that the amount of HIV-1 Gag-specific mRNA (Fig. 6a) produced in SLPI overexpressed monkey and human cells transduced with HIV-1 molecular clone pNL4-3 was three- and six-fold lower, respectively, compared to those in which SLPI was not over-expressed (Fig. 6a). Reduction of Gag mRNA expression in the presence of SLPI in CV1 cells was lower compared to 293T cells due to the already low levels of Gag mRNA in CV1 cells transduced with pNL4-3 plasmid. This could be explained due to increased overexpression of SLPI in the presence of HIV-1 Tat protein produced in pNL4-3 transduced CV1 cells (Fig. 6b) and confirms our earlier findings (Fig. 5). These results hint to an intracellular inhibitory effect of SLPI on HIV-1 replication, which could not be due to the known extracellular activity of SLPI, since we infected cells with an HIV-1 molecular clone by transfection rather than using infectious viral particles, bypassing virus attachment and entry steps. Also, since the infections of CV-1 and 293T cells were single-round due to the lack of cellular receptor, CD4, for HIV-1 [31] in these cells [21, 22], the possibility of SLPI’s extracellular inhibitory effect on virus entry [11, 12] to initiate new infections, especially in culture medium of 293T cells was excluded.
Since our findings hint to an intracellular anti-HIV-1 activity of SLPI in addition to its previously known extracellular anti-HIV-1 activity, we speculate that, in cases where HIV-1 particles escape from restriction factors and manage to infect monkey cells, induction of SLPI by Tat can lead to lower virus production in infected cells. We also speculate that since HIV-1 Tat protein secreted by infected cells is known to diffuse into neighboring cells [32], it may induce an anti-HIV-1 state in uninfected cells by inducing SLPI production and secretion and thus protecting neighboring cells from HIV-1 infection through SLPI’s extracellular anti-HIV-1 activity [11, 12]. Thus, overexpression of SLPI in HIV-1 infected cells could potentially tip the balance in favor of the host immune system in a fight against HIV-1 infection and lead to the elimination of the virus from the host.
SLPI is also known to have anti-inflammatory effects through its prevention of the activation of nuclear factor kB (NF-kB), a transcription factor that increases the synthesis of several pro-inflammatory mediators [20, 33, 34]. Thus, it may have an extra role in protecting an HIV-1 infected host against progression into AIDS. In many studies, it has been shown that persistent activation of the immune system (chronic inflammation) during HIV-1 infection is the most important cause of T cell reduction and progression to AIDS [35,36,37,38]. In a study performed in AGMs, it was determined that the anti-inflammatory profile developed in SIV infected monkeys protected them against AIDS [39]. Anti-inflammatory profile in SIV infected monkeys was due to an excessive increase in anti-inflammatory cytokine IL-10 levels and stable levels of pro-inflammatory cytokines. In a different study, it was shown that SLPI caused overproduction of IL-10 in human macrophage cells [40], suggesting a potential role of SLPI in the development of anti-inflammatory profile in humans and contribution to delay in AIDS progression through IL-10. These data suggest that in addition to reducing HIV-1 production in infected cells, SLPI may also have a preventive role in progression to AIDS in infected individuals. A study in Colombia, on elite controllers of HIV-1 infection, showed that PBMC cells in those patients produced significantly higher amounts of SLPI compared to non-controller patients [41]. Also, the immune system activation in elite controllers remained low compared to non-controllers, supporting our argument.
Our results suggest the presence of a previously undiscovered anti-HIV-1 mechanism mediated by SLPI in monkey cells. In addition, the increase of SLPI production by AGM cells in the presence of HIV-1 Tat protein indicates that these cells evolved to detect and respond to HIV-1 infection. We believe that the investigation of the subject in different AGM and other OWM cells known to be resistant to HIV-1 is essential to reinforce our notion and to lead to future clinical studies using SLPI for the control of HIV-1 infection and AIDS progression in patients infected with HIV-1.
References
McCarthy KR, Kirmaier A, Autissier P, Johnson WE (2015) Evolutionary and functional analysis of old world primate TRIM5 reveals the ancient emergence of primate lentiviruses and convergent evolution targeting a conserved capsid interface. PLoS Pathog. https://doi.org/10.1371/journal.ppat.1005085
Klatt NR, Silvestri G, Hirsch V (2012) Nonpathogenic simian immunodeficiency virus infections. Cold Spring Harb Perspect Med. https://doi.org/10.1101/cshperspect.a007153
Pepin J (2013) The origins of AIDS: from patient zero to ground zero. J Epidemiol Community Health 67(6):473–475. https://doi.org/10.1136/jech-2012-201423
Blanco-Melo D, Venkatesh S, Bieniasz PD (2012) Intrinsic cellular defenses against human immunodeficiency viruses. Immunity 37(3):399–411. https://doi.org/10.1016/j.immuni.2012.08.013
Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J (2004) The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 427(6977):848–853. https://doi.org/10.1038/nature02343
Mariani R, Chen D, Schrofelbauer B, Navarro F, Konig R, Bollman B, Munk C, Nymark-McMahon H, Landau NR (2003) Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell 114(1):21–31. https://doi.org/10.1016/S0092-8674(03)00515-4
Saito A, Akari H (2013) Macaque-tropic human immunodeficiency virus type 1: breaking out of the host restriction factors. Front Microbiol. https://doi.org/10.3389/fmicb.2013.00187
Hatziioannou T, Del Prete GQ, Keele BF, Estes JD, McNatt MW, Bitzegeio J, Raymond A, Rodriguez A, Schmidt F, Mac Trubey C, Smedley J, Piatak M, KewalRamani VN, Lifson JD, Bieniasz PD (2014) HIV-1-induced AIDS in monkeys. Science 344(6190):1401–1405. https://doi.org/10.1126/science.1250761
de la Fuente C, Santiago F, Deng LW, Eadie C, Zilberman I, Kehn K, Maddukuri A, Baylor S, Wu KL, Lee CG, Pumfery A, Kashanchi F (2002) Gene expression profile of HIV-1 Tat expressing cells: a close interplay between proliferative and differentiation signals. BMC Biochem. https://doi.org/10.1186/1472-2091-3-14
McNeely TB, Dealy M, Dripps DJ, Orenstein JM, Eisenberg SP, Wahl SM (1995) Secretory leukocyte protease inhibitor: a human saliva protein exhibiting anti-human immunodeficiency virus 1 activity in vitro. J Clin Investig 96(1):456–464. https://doi.org/10.1172/JCI118056
McNeely TB, Shugars DC, Rosendahl M, Tucker C, Eisenberg SP, Wahl SM (1997) Inhibition of human immunodeficiency virus type 1 infectivity by secretory leukocyte protease inhibitor occurs prior to viral reverse transcription. Blood 90(3):1141–1149
Py B, Basmaciogullari S, Bouchet J, Zarka M, Moura IC, Benhamou M, Monteiro RC, Hocini H, Madrid R, Benichou S (2009) The phospholipid scramblases 1 and 4 are cellular receptors for the secretory leukocyte protease inhibitor and interact with CD4 at the plasma membrane. PLoS ONE. https://doi.org/10.1371/journal.pone.0005006
Kuhmann SE, Platt EJ, Kozak SL, Kabat D (2000) Cooperation of multiple CCR5 coreceptors is required for infections by human immunodeficiency virus type 1. J Virol 74(15):7005–7015. https://doi.org/10.1128/Jvi.74.15.7005-7015.2000
Jensen FC, Girardi AJ, Gilden RV, Koprowski H (1964) Infection of human and simian tissue cultures with rous sarcoma virus. Proc Natl Acad Sci USA 52:53–59
Osada N, Kohara A, Yamaji T, Hirayama N, Kasai F, Sekizuka T, Kuroda M, Hanada K (2014) The genome landscape of the African green monkey kidney-derived vero cell line. DNA Res 21(6):673–683. https://doi.org/10.1093/dnares/dsu029
Dubridge RB, Tang P, Hsia HC, Leong PM, Miller JH, Calos MP (1987) Analysis of mutation in human-cells by using an epstein-barr-virus shuttle system. Mol Cell Biol 7(1):379–387. https://doi.org/10.1128/Mcb.7.1.379
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods 25(4):402–408. https://doi.org/10.1006/meth.2001.1262
Kondo M, Sudo K, Tanaka R, Sano T, Sagara H, Iwamuro S, Takebe Y, Imai M, Kato S (2009) Quantitation of HIV-1 group M proviral DNA using TaqMan MGB real-time PCR. J Virol Methods 157(2):141–146. https://doi.org/10.1016/j.jviromet.2008.12.006
Vroling AB, Konijn T, Samsom JN, Kraal G (2011) The production of secretory leukocyte protease inhibitor by dendritic cells. Mol Immunol 48(4):630–636. https://doi.org/10.1016/j.molimm.2010.11.002
Scott A, Weldon S, Taggart CC (2011) SLPI and elafin: multifunctional antiproteases of the WFDC family. Biochem Soc T 39(5):1437–1440. https://doi.org/10.1042/BST0391437
Geijtenbeek TBH, Kwon DS, Torensma R, van Vliet SJ, van Duijnhoven GCF, Middel J, Cornelissen ILMHA, Nottet HSLM, KewalRamani VN, Littman DR, Figdor CG, van Kooyk Y (2000) DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell 100(5):587–597. https://doi.org/10.1016/S0092-8674(00)80694-7
Ndung'u T, Renjifo B, Novitsky VA, McLane MF, Gaolekwe S, Essex M (2000) Molecular cloning and biological characterization of full-length HIV-1 subtype C from Botswana. Virology 278(2):390–399. https://doi.org/10.1006/viro.2000.0583
Coren LV, Trivett MT, Jain S, Ayala VI, Del Prete GQ, Ohlen C, Ott DE (2015) Potent restriction of HIV-1 and SIVmac239 replication by African Green Monkey TRIM5 alpha. Retrovirology. https://doi.org/10.1186/s12977-015-0137-9
Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA (1986) Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol 59(2):284–291
Igarashi T, Iyengar R, Byrum RA, Buckler-White A, Dewar RL, Buckler CE, Lane HC, Kamada K, Adachi A, Martin MA (2007) Human immunodeficiency virus type 1 derivative with 7% simian immunodeficiency virus genetic content is able to establish infections in pig-tailed macaques. J Virol 81(20):11549–11552. https://doi.org/10.1128/Jvi.00960-07
Saito A, Nomaguchi M, Iijima S, Kuroishi A, Yoshida T, Lee YJ, Hayakawa T, Kono K, Nakayama EE, Shioda T, Yasutomi Y, Adachi A, Matano T, Akari H (2011) Improved capacity of a monkey-tropic HIV-1 derivative to replicate in cynomolgus monkeys with minimal modifications. Microbes Infect 13(1):58–64. https://doi.org/10.1016/j.micinf.2010.10.001
Clark E, Nava B, Caputi M (2017) Tat is a multifunctional viral protein that modulates cellular gene expression and functions. Oncotarget 8(16):27569–27581. https://doi.org/10.18632/oncotarget.15174
Cantalupo PG, Saenz-Robles MT, Rathi AV, Beerman RW, Patterson WH, Whitehead RH, Pipas JM (2009) Cell-type specific regulation of gene expression by simian virus 40 T antigens. Virology 386(1):183–191. https://doi.org/10.1016/j.virol.2008.12.038
Li MM, MacDonald MR, Rice CM (2015) To translate, or not to translate: viral and host mRNA regulation by interferon-stimulated genes. Trends Cell Biol 25(6):320–329. https://doi.org/10.1016/j.tcb.2015.02.001
Jana NK, Gray LR, Shugars DC (2005) Human immunodeficiency virus type 1 stimulates the expression and production of secretory leukocyte protease inhibitor (SLPI) in oral epithelial cells: a role for SLPI in innate mucosal immunity. J Virol 79(10):6432–6440. https://doi.org/10.1128/JVI.79.10.6432-6440.2005
LaBonte JA, Babcock GJ, Patel T, Sodroski J (2002) Blockade of HIV-1 infection of new world monkey cells occurs primarily at the stage of virus entry. J Exp Med 196(4):431–445. https://doi.org/10.1084/jem.20020468
Frankel AD, Pabo CO (1988) Cellular uptake of the tat protein from human immunodeficiency virus. Cell 55(6):1189–1193. https://doi.org/10.1016/0092-8674(88)90263-2
Henriksen PA, Hitt M, Xing Z, Wang J, Haslett C, Riemersma RA, Webb DJ, Kotelevtsev YV, Sallenave JM (2004) Adenoviral gene delivery of elafin and secretory leukocyte protease inhibitor attenuates NF-kappa B-dependent inflammatory responses of human endothelial cells and macrophages to atherogenic stimuli. J Immunol 172(7):4535–4544. https://doi.org/10.4049/jimmunol.172.7.4535
Moreau T, Baranger K, Dade S, Dallet-Choisy S, Guyotl N, Zani ML (2008) Multifaceted roles of human elafin and secretory leukocyte proteinase inhibitor (SLPI), two serine protease inhibitors of the chelonianin family. Biochimie 90(2):284–295. https://doi.org/10.1016/j.biochi.2007.09.007
Sousa AE, Carneiro J, Meier-Schellersheim M, Grossman Z, Victorino RMM (2002) CD4 T cell depletion is linked directly to immune activation in the pathogenesis of HIV-1 and HIV-2 but only indirectly to the viral load. J Immunol 169(6):3400–3406. https://doi.org/10.4049/jimmunol.169.6.3400
Hazenberg MD, Otto SA, van Benthem BHB, Roos MTL, Coutinho RA, Lange JMA, Hamann D, Prins M, Miedema F (2003) Persistent immune activation in HIV-1 infection is associated with progression to AIDS. AIDS 17(13):1881–1888. https://doi.org/10.1097/01.aids.0000076311.76477.6e
Deeks SG, Kitchen CM, Liu L, Guo H, Gascon R, Narvaez AB, Hunt P, Martin JN, Kahn JO, Levy J, McGrath MS, Hecht FM (2004) Immune activation set point during early HIV infection predicts subsequent CD4+ T-cell changes independent of viral load. Blood 104(4):942–947. https://doi.org/10.1182/blood-2003-09-3333
van Asten L, Danisman F, Otto SA, Borghans JAM, Hazenberg MD, Coutinho RA, Prins M, Miedema F (2004) Pre-seroconversion immune status predicts the rate of CD4 T cell decline following HIV infection. AIDS 18(14):1885–1893. https://doi.org/10.1097/00002030-200409240-00004
Kornfeld C, Ploquin MJY, Pandrea I, Faye A, Onanga R, Apetrei C, Poaty-Mavoungou V, Rouquet P, Estaquier J, Mortara L, Desoutter JF, Butor C, Le Grand R, Roques P, Simon F, Barre-Sinoussi F, Diop OM, Muller-Trutwin MC (2005) Antiinflammatory profiles during primary SIV infection in African green monkeys are associated with protection against AIDS. J Clin Investig 115(4):1082–1091. https://doi.org/10.1172/Jci200523006
Sano C, Shimizu T, Sato K, Kawauchi H, Tomioka H (2000) Effects of secretory leucocyte protease inhibitor on the production of the anti-inflammatory cytokines, IL-10 and transforming growth factor-beta (TGF-beta), by lipopolysaccharide-stimulated macrophages. Clin Exp Immunol 121(1):77–85. https://doi.org/10.1046/j.1365-2249.2000.01269.x
Taborda NA, Catano JC, Delgado JC, Rugeles MT, Montoya CJ (2012) Higher SLPI expression, lower immune activation, and increased frequency of immune cells in a cohort of Colombian HIV-1 controllers. JAIDS-J Acq Imm Def 60(1):12–19. https://doi.org/10.1097/QAI.0b013e31824876ca
Acknowledgements
The authors would like to thank to the National Biological Mass Spectrometry and Proteomics Facility, and Biotechnology and Bioengineering Research and Application Centers of Izmir Institute of Technology for their help and technical support.
Funding
The research was funded by The Scientific and Technological Research Council of Turkey (TUBITAK), Grant No.: 115S213.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by AA, SÖ and BŞ. The first draft of the manuscript was written by AA and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Research involving human and animal participants
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Edited by Wolfram H. Gerlich.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Özdemir, S., Şengez, B. & Arslanoğlu, A. Human immunodeficiency virus type-1 Tat protein induces secretory leukocyte protease inhibitor expression in African green monkey but not human cells. Virus Genes 56, 182–193 (2020). https://doi.org/10.1007/s11262-020-01731-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-020-01731-x