
Abstract
A novel gamma-retroviral sequence (7912 bp), inclusive of both partial 5′ and 3′ long terminal repeat regions, was identified from the brain of a black flying-fox (Pteropus alecto), Queensland, Australia. The sequence was distinct from other retroviral sequences identified in bats and showed greater identity to Koala, Gibbon ape leukaemia, Melomys burtoni and Woolly monkey retroviruses, forming their own phylogenetic clade. This finding suggests that these retroviruses may have an unknown common ancestor and that further investigation into the diversity of gamma-retroviruses in Australian Pteropus species may elucidate their evolutionary origins.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Retroviruses are known to cause leukaemia, lymphomas, neurological disease and immuno-deficiencies in a wide range of species [1]. The Retroviridae family is divided into two subfamilies, the Spumaretrovirinae and the Orthoretrovirinae, the latter comprising six genera, inclusive of the gamma-retroviruses [2]. All retroviruses have a replication strategy that involves the conversion of viral RNA into double-stranded DNA which is inserted into the host’s genome creating a provirus [1]. Infectious exogenous retroviruses are transmitted horizontally or vertically between hosts, whereas endogenous retroviruses, which are integrated into germline cells, are inherited. Endogenous retroviruses are usually defective, but can be full-length and may have the ability to give rise to exogenous viruses [2]. While gamma-retroviruses have been found in many mammalian species, their evolutionary origins are unknown [3].
Koala retrovirus (KoRV) was fully characterized as an intact gamma-retrovirus of Australian koalas by Hanger et al. in 2000 [4], and found to be endogenous in the genome of northern Australian koala populations [5, 6]. However, as some koala populations in southern Australia appear free of KoRV and KoRV-positive animals demonstrate high levels of viral replication, it appeared that KoRV may be an active exogenous retrovirus currently undergoing a natural process of endogenisation, an interesting evolutionary event as a retrovirus invades a mammalian genome [5]. KoRV viral sequences are closely related to those of the exogenous gamma-retrovirus Gibbon ape leukaemia virus (GALV), a retrovirus isolated from captive gibbons in Thailand [4]. It has been suggested that these two viruses may have been transferred to their hosts through separate host switching events, as there is no evidence to suggest that KoRV and GALV are recombinants or have co-evolved [7, 8]. A direct species jump seems geographically improbable, but transmission to both species from a common host that is a reservoir for an ancestral virus is more plausible [9].
Retroviral sequences with a close genetic relationship to GALV have been discovered in an Australian rodent species Melomys burtoni [9] and the Indonesian sub-species of Melomys burtoni [10], named MbRV and MelWMV, respectively. This finding is interesting in the context of the relationship between KoRV and GALV considering the host biogeography and the potential role of rodents as vectors for interspecies viral transmission of retroviruses [2]. Previously, several gamma-retroviruses have been isolated from bat species that have basal phylogenetic ancestries for gamma-retroviruses [3, 11], but none have been established as closely related to either KoRV or GALV. This study presents a novel bat gamma-retroviral sequence that is related to KoRV, GALV, MelWMV, MbRV and Woolley monkey retrovirus (WMV), potentially sharing a common ancestor.
A black flying-fox (Pteropus alecto) was submitted to the Biosecurity Sciences Laboratory (BSL), Queensland, Australia, in July 2017 for investigation by next-generation sequencing (NGS). Total RNA was extracted from the flying-fox brain sample using the RNeasy Mini Kit (Qiagen), an on-column DNAase digestion performed following manufacturer’s instructions, with a final elution volume of 30 µl. RNA concentration was measured using the Qubit Fluorometer and the Qubit RNA HS Assay Kit (ThermoFisher Scientific). The sequence library was prepared using the TruSeq Stranded mRNA Library Preparation Kit substituting the Oligo-dT capture beads with those from the Ribo-Zero™ rRNA Removal Kit (Human/Mouse/Rat, Illumina) to deplete the ribosomal RNA. cDNA was prepared using Superscript™ II Reverse Transcriptase (ThermoFisher Scientific) and all purification steps utilized AMPure XP kit paramagnetic beads (Beckman Coulter). The size and purity of the pooled sequence library were quantified using the 2200 TapeStation (Agilent), with the final equimolar concentration quantified using the Qubit dsDNA HS Assay Kit (ThermoFisher Scientific). The library was sequenced on a NextSeq 500 Sequencing Platform using a NextSeqMid Output Kit v2 300 (Illumina).
Indexing quality control and FASTQ file generation were initially performed using the online server BaseSpace Sequence Hub (Illumina), with additional trimming performed using the Geneious® version 11.0.3 (http://www.geneious.com [12]) (Biomatters) plugin BBDuk (Brian Bushnell). Continuing with the Geneious® assembler, de novo assembly was performed on the samples 12.5 million paired reads and sequence homology searches of the resulting contigs performed using GenBanks (NCBI) basic local alignment search tool (BLAST) against the CoreNucleotide collection [13] (http://www.ncbi.nlm.nih.gov/genbank). Additional direct Sanger sequencing was conducted using primers specific to the NGS spanning areas of low reads. A consensus sequence for a novel flying-fox retrovirus (FFRV) was exported and submitted to Genbank (Accession: MK040728). Iterative mapping (5 times) against the final consensus sequence was performed using Geneious® but did not acquire any additional reads.
Analysis identified a novel full-length gamma-retroviral sequence inclusive of both partial 5′ and 3′ long terminal repeat regions (7912 bp). The FFRV sequence was distinct from other gamma-retroviral sequences found in bat species with only 48% identity to the full-length sequence of Rhinolophus ferrumequinum retrovirus (RfRV: JQ303225) in pairwise comparison. Only the pol genes from other bat retroviruses were available for comparison, where identity was again low: Pteropus alecto retrovirus (PaRV: JQ292910, 56%), Rousettus leschenaultii retrovirus (RlRV: JQ951958, 61%) and Megaderma lyra retrovirus (MlRV: JQ951956, 66%). The FFRV sequence showed greater identity with the full-length sequences of KoRV (NC039228, 74%), GALV (NC001885, 79%) and WMV (KT724051, 81%) and formed a phylogenetic clade that included MelWMV (KX059700, 81% identity). Only partial MbRV pol gene (KF572484) was available for comparison against FFRV, and identity was also high (83%).
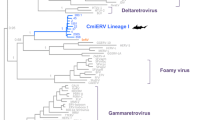
To characterize the evolutionary relationship between FFRV and other gamma-retroviruses, full-length retroviral sequences were aligned in Geneious® using Muscle [14] and phylogenetic trees were built using model selection tools in PhyML [15]. The general time reversible substitution model was employed and the bootstrapped (n = 1000) maximum likelihood phylogenetic tree is presented in Fig. 1. To estimate the relationships between FFRV and other retroviruses at the protein level, phylogenetic trees were built using the model selection method described by Hall et al. [16]. Seventeen sequences homologous to the FFRV Pol gene (3449 bp) were identified using BLAST, aligned using Muscle and translated into amino acid sequences. The Jones–Taylor–Thornton amino acid substitution model [17] was employed to best describe the pattern using a discrete Gamma distribution (JTT + G), and the maximum likelihood tree for the retroviral Pol genes is presented in Fig. 2.

Molecular phylogenetic analysis of the complete retroviral genome of the gamma-retroviruses, using the maximum likelihood method based on the general time reversible substitution model. The tree with the highest log likelihood is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. (Reticuloendotheliosis virus REV: NC006934; Rhinolophus ferrumequinum retrovirus RfRV: JQ303225; Feline leukaemia virus FLV: NC001940; Moloney murine leukaemia virus MLMCV: NC001501; Gibbon ape leukaemia virus GALV: NC001885; Woolly monkey virus WMV: Melomys woolly monkey virus MelWMV: KX059700; Koala retrovirus KoRV NC039228; Mus caroli endogenous virus McERV: KC460271; Mus dunni endogenous virus MDEV: AF053745; Porcine endogenous retrovirus PERV: AF038600; Flying-fox retrovirus FFRV: MK040728)

Molecular phylogenetic analysis of the Pol gene amino acid sequence of the KoRV/GALV/WMV/FFRV clade, using the maximum likelihood method based on the JTT matrix-based model. The tree with the highest log likelihood is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches
The finding of a flying-fox gamma-retrovirus forming a clade with KoRV, GALV and WMV (Fig. 2) informs investigations into the evolutionary origins of these retroviruses. It is not surprising to identify a novel gamma-retrovirus in an Australian Pteropus species as Chiropteran species (bats) are known reservoirs for many viruses [3]. Our finding supports the suggestion of Ciu et al. [3] that there may be several origins of retroviruses in bat species, and that indeed bats may be hosts for diverse retroviruses. This is evidenced by the FFRV being quite distinct from both PaRV from the same species of flying-fox (P. alecto) and retroviruses from other bat species (Rousettus leschenaultia, Megaderma lyra, Rhinolophus ferrumequinum). While FFRV does exhibit three functional ORFs (gag, pol and env genes), the presence of partial LTRs is expected as retroviral LTRs are only partially transcribed into an RNA intermediate. This is suggestive of the virus being endogenous in nature. However, the exogenous versus endogenous nature of this novel virus will be the focus of future epidemiological investigation of FFRV in Australian Pteropus species.
As suggested by Simmons et al. [9] and Greenwood et al. [2], with respect to the hypothesis of “species jumping” of retroviruses closely related to GALV and KoRV, the most likely candidates are those species that transit between the Australian mainland and South East Asia, with geographic ranges and feeding ecology that may result in close contact with both gibbons and koalas. Ciu et al. [3, 18] suggested that bats that harboured distinct gamma-retroviruses may have played an important role as reservoir hosts during the diversification of mammalian gamma-retroviruses, and that bat retroviruses are not constrained by geographic barriers. Denner [19] similarly suggests the hypothesis that retroviruses of bats are the origin of GaLV and KoRV which also deserve consideration. Alternatively, McKee et al. [20] suggest that Melomys burtoni retrovirus would be a prime candidate for such an ancestral virus.
Notwithstanding these hypotheses, the relationship between KoRV, GALV, MelWMV, MbRV, WMV and FFRV is still unclear. From our phylogenetic analysis, it may be that the ancestor of all these retroviruses infected an unknown host, or alternatively, these retroviruses may have an unknown common ancestor. From our phylogenetic analyses, the pattern is compatible with all possible host-gamma-retrovirus co-divergence versus species switch scenarios. Thus, further investigation into the diversity of gamma-retroviruses in Australian Pteropus species may elucidate their evolutionary origins.
References
Coffin JM, Hughes SH, Varmus HE (1997) The interactions of retroviruses and their hosts. In: Coffin JM, Hughes SH, Varmus HE (eds) Retroviruses. Cold Spring Harbor Laboratory Press, New York, p 335–342
Greenwood AD, Ishida Y, O’Brien SP, Roca AL, Eiden MV (2018) Transmission, evolution and endogenization: lessons learned from recent retroviral invasions. Micro Mol Biol Rev 82:1
Cui J, Tachedjian M, Wang L, Tachedjian G, Wang LF, Zhanga S (2012) Discovery of retroviral homologs in bats: implications for the origin of mammalian gammaretroviruses. J Virol 86(8):4288–4293
Hanger J, Bromham L, McKee J, O’Brien T, Robinson W (2000) The nucleotide sequence of koala (Phascolarctos cinereus) retrovirus: a novel type C endogenous virus related to Gibbon ape leukemia virus. J Virol 74:4264–4272
Tarlinton R, Meers J, Young P (2006) Retroviral invasion of the koala genome. Nature 442:79–81
Ishida Y, Zhao K, Greenwood A, Roca A (2015) Proliferation of endogenous retroviruses in the early stages of a host germ line invasion. Mol Biol Evol 32:109–120
Bromham L (2002) The human zoo: endogenous retroviruses in the human genome. Trends Ecol Evol 17:91–97
Stoye J (2006) Koala retrovirus: a genome invasion in real time. Genome Biol 7:241
Simmons G, Clarke D, McKee J, Young P, Meers J (2014) Discovery of a novel retrovirus sequence in an Australian native rodent (Melomys burtoni): a putative link between Gibbon ape leukemia virus and koala retrovirus. PLoS ONE 9(9)
Alfano N, Michaux J, Morand S, Aplin K, Tsangaras K, Löber U, Fabre P, Fitriana Y, Semiadi G, Ishida Y, Helgen K, Roca A, Eiden M, Greenwood A (2016) Endogenous Gibbon ape leukemia virus identified in a rodent (Melomys burtoni subsp.) from Wallacea (Indonesia). Virol 90(18):8169–8180
Yuan L, Li M, Li L, Monagin C, Chmura A, Schneider B, Epstein J, Mei X, Shi Z, Daszak P, Chen J (2014) Evidence for retrovirus and paramyxovirus infection of multiple bat species in China. Viruses 6:2138–2154
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Mentjies P, Drummond A (2012) Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28(12):1647–1649
Genbank. http://www.ncbi.nlm.nih.gov. Accessed Sept 2018
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32(5):1792–1797
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59(3):307–321
Hall BG (2013) Building phylogenetic trees from molecular data with MEGA. Mol Biol Evol 30(5):1229–1235
Jones DT, Taylor WR, Thornton JM (1992) The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci 8:275–282
Cui J, Tachedjian G, Tachedjian M, Holmes E, Zhang S, Wang L (2012) Identification of diverse groups of endogenous gammaretroviruses in mega- and microbats. J Gen Virol 93:2037–2045
Denner J (2016) Transspecies transmission of gammaretroviruses and the origin of the Gibbon ape leukaemia virus (GaLV) and the Koala retrovirus (KoRV). Viruses 8:336
McKee J, Clark N, Shapter F, Simmons G (2017) A new look at the origins of Gibbon ape leukemia virus. Virus Genes 53:165–172
Acknowledgements
The authors would like to thank Dr. Gavin Wilkie, Ali Hoeger and the management and staff of the Biosecurity Sciences Laboratory for technical assistance and their generous support; Dr. Gregory Simmons for discussions; and Dr. Janine Barrett and Dr. Darren Underwood for reviewing the manuscript. The project was funded by Biosecurity Queensland, the University of Queensland and Wildlife Health Australia.
Author information
Authors and Affiliations
Contributions
All authors have reviewed the final version of the manuscript and approved it for publication. Conceived of or designed the study: LM, CS, AG, KA, JM; performed research: LM, CS; analysed data: LM, CS, JO; wrote/reviewed the paper: LM, CS, AG, KA, JM, JO.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
All authors have reviewed the final version of the manuscript and approve it for publication.
Additional information
Edited by Dr. Juergen A Richt.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
McMichael, L., Smith, C., Gordon, A. et al. A novel Australian flying-fox retrovirus shares an evolutionary ancestor with Koala, Gibbon and Melomys gamma-retroviruses. Virus Genes 55, 421–424 (2019). https://doi.org/10.1007/s11262-019-01653-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-019-01653-3