
Abstract
Noroviruses are leading cause of acute gastroenteritis worldwide. In our previous study, we established an in vitro histo-blood group antigens (HBGAs) binding blockade assay against GII.3 Norovirus virus like particles (VLPs) with trypsin digestion. In this study, we characterized the blocking antibody binding site and epitope type (linear or conformational) by using hyperimmune sera produced against different antigens. VP1 from Jingzhou402 (GII.3, JZ402) strain was expressed by using pGEX-6p-1 expression vector and the insoluble proteins were purified for immunization in rabbit. Previously characterized chimeric VP1-assembled VLPs (GII.4-VP1/GII.3-P2) were used to immunize guinea pig. Peptides reactive with hyperimmune serum against VLPs derived from the VP1 of JZ402 strain were conjugated with BSA and used to immunize rabbits. Hyperimmune sera against above antigens and JZ402 and JZ403 strain-derived VLPs were used to compare their HBGAs blocking effects. Rabbit anti-GST-VP1 and BSA-peptide conjugated hyperimmune sera demonstrated no blocking effects against the binding of GII.3 and GII.4 NoV VLPs to salivary HBGAs. Guinea pig anti-GII.4-VP1/GII.3-P2 hyperimmune serum blocked the binding of trypsin cleaved GII.3 VLPs to salivary HBGAs with no or very weak blocking effects against the binding of GII.4 VLPs to salivary HBGAs. Our data indicated that HBGAs blocking antibodies primarily bound the P2 domain of GII.3 NoV VP1 and their binding epitopes were most probably conformation-dependent.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Noroviruses (NoVs), members of the family Caliciviridae, are non-enveloped, single-stranded, and positive-sense RNA viruses with genome sizes of about 7.5–7.7 kb excluding the polyadenylation tail. The genome of human NoVs is organized in the order of 5′ end short untranslated region, open reading frame 1 (ORF1), ORF2, ORF3, and 3′ end untranlated region. ORF1 encodes a polyprotein that is further cleaved into six non-structural proteins. ORF2 and 3 encode major capsid protein VP1 and minor capsid protein VP2, respectively. VP1 can be structurally divided into shell (S) domain and protruding (P) domain with the latter being further divided into P1 and P2 domains [1, 2]. Amino acid sequence in the P2 domain is highly variable, which is related with different binding patterns against histo-blood group antigens (HBGAs), the presumed receptor, or co-receptor for NoVs. Based on amino acid sequences of the VP1, NoVs can be divided at least into seven genogroups namely GI through to GVII, with GI, GII, and GIV infecting humans [3]. NoVs in GI and GII can be further divided into more than 30 genotypes.
The HBGA binding sites have been extensively characterized for the VP1 of Norwalk (GI.1) and GII.4 NoVs by epitope mapping and crystallography technique [4,5,6,7,8,9,10]. Due to weak or absence of binding of expressed GII.3 NoV VLPs to synthetic or salivary HBGAs, no specific binding epitopes (amino acids involved in binding with HBGAs) have been determined so far [11,12,13]. In our previous study, we found that trypsin digestion significantly promoted the binding of GII.3 NoV VLPs to salivary HBGAs and the enhancement may be due to formation of polymers from cleaved VP1. Based on this finding, we successfully established an in vitro HBGA binding blockade assay, which provides us an opportunity to characterize regions or amino acids that are involved in binding with HBGAs [14].
To characterize the epitopes recognized by HBGA blocking antibodies, in this study, we first expressed the full-length VP1 of a GII.3 strain (Jingzhou402, Genbank accession number, KF306213) by using prokaryotic expression vector pGEX-6p-1 and the expressed glutathione-S-transferase-fused protein (GST-VP1) was purified and used for immunization of rabbits. Previously characterized chimeric VP1 (GII.4-VP1/GII.3-P2)-assembled VLPs with P2 domain from the Jingzhou402 strain (here designated as JZ402) in the backbone of a Jingzhou403 (GII.4) strain (Genbank accession number, KF306213) were purified and used to immunize guinea pigs [15]. Peptides reactive with hyperimmune sera against VLPs derived from JZ402 strain were conjugated with BSA and used to immunize rabbits. Hyperimmune sera against above antigens were used to characterize their blocking effects over the binding of GII.3 (Genbank accession number KY767665, here designated as ZZ3-34) and GII.4 VLPs (here designated as JZ403) to salivary HBGAs.
Materials and methods
Cloning, expression, and purification of GST-VP1
To express the fusion protein GST-VP1, the VP1 coding sequence of the Jingzhou402 strain was amplified by forward primer (FP) with BamHI restriction enzyme site and reverse primer (RP) with NotI restriction enzyme site: FP, GGATCCATGAAGATGGCGTCGAATGA; RF, TAAAGCGGCCGCTTATTGAATCCTTCTACGCC. The amplified fragment was ligated into pGEX-6p-1 expression vector. The construct was sequenced to confirm correct insertion before used for following transformation of BL21 (DE3) competent cells. Transformed cell which formed colonies under selection with antibiotic ampicillin were picked for colony-PCR for identification of positive clones with the above-mentioned FP and RP set.
Positive clones were incubated at 37 °C till the optical density (OD) value at 600 nm reached 0.6–0.8 and then isopropyl β-d-thiogalactoside (IPTG) was added to a final concentration of 0.1 mM. Bacteria without addition of IPTG were used as control. The bacteria were further cultured at 37 °C for 6 h before harvest. Harvested cells were pelleted, washed with ice-cold phosphate buffered saline (PBS, 0.01 M, pH 7.2), sonicated, and pelleted again. The supernatant and pellet were analyzed by SDS-PAGE to confirm the solubility of expressed fusion protein, GST-VP1. GST-VP1 was expressed as inclusion body. The insoluble GST-VP1 was then purified by gel separation and electroelution. Briefly, the cell pellet containing insoluble GST-VP1 was washed with ice-cold PBS for five times before boiled in loading buffer containing 1% SDS, 1% 2-mercaptoethanol, 0.25 M Tris–HCl buffer (pH 6.8), 10% glycerol, and 0.0025% bromophenol blue. Boiled proteins were loaded into SDS-PAGE gel consisting of 3% concentration gel and 10% separating gel and separated by using Hoefer SE600-15-1.5 Vertical Electrophoresis System. The gel was stained with water dissolved Coomassie blue and then destained with distilled water till target protein was visible. The target protein in the gel was cut out and recovered by electroelution with 422 Electro-Eluter according to the manufacturer’s instructions. The eluted protein was dialyzed in Slide-A-Lyzer™ Dialysis Cassette with a molecular weight cut-off value of 10 kDa to remove SDS. The concentration of finally dialyzed protein was determined by SDS-PAGE with bovine serum albumin (BSA) as standards.
Purification of NoV VLPs
The purification procedures for ZZ3-34 and JZ403 NoV VLPs, as well as chimeric protein GII.4-VP1/GII.3-P2-assembled VLPs have been described previously [14, 15]. Briefly, VLPs in sf9 cell culture medium were pelleted by ultracentrifugation and purified by cesium chloride density gradient centrifugation. The target band was collected and further ultracentrifuged to remove cesium chloride. The final protein concentration was determined by using Nanodrop 2000. The purity of purified proteins was analyzed by SDS-PAGE and the morphology of assembled VLPs was observed under electron microscopy (EM).
Production of hyperimmune sera
To prepare hyperimmune sera against fusion protein GST-VP1 and chimeric protein GII.4-VP1/GII.3-P2, both proteins were used to immunize rabbits and guinea pig, respectively. The procedure for preparation of hyperimmune serum in rabbits was performed as described previously [12]. Briefly, purified fusion protein GST-VP1 was used to immunize two Japan big ear rabbits subcutaneously at a 2-week intervals for a total of four immunizations at 500 µg/dose mixed at 1:1 ratio with Freund’s complete adjuvant (first immunization) or Freund’s incomplete adjuvant (subsequent immunizations). For guinea pig immunization, the same immunization protocol was used except that a dose of 50 µg was used. Sera were collected from rabbit cervical artery under anesthesia and direct heart puncture for guinea pig 1 week after the last immunization and anti-VLP-specific IgG titers were determined by using indirect enzyme-linked immunosorbent assay (ELISA). GST-VP1, GII.4-VP1/GII.3-P2-assembled VLPs, and previously characterized ZZ3-34 and JZ403 NoV VLPs were used to determine the genotype-specific and cross-genotype IgG titers. Briefly, GST-VP1, ZZ3-34, JZ403, and chimeric protein GII.4-VP1/GII.3-P2-assembled VLPs at 0.5 µg/ml in carbonate buffer (pH 9.6) were coated onto microplate wells. The plate was blocked with PBS containing 0.25% Tween-20 (PBS-T) and 1% BSA and collected hyperimmune serum was 10-fold serially diluted in PBS-T and added to above wells. The bound antibodies were detected by horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG and optical density (OD) value at 450 nm was measured by using a Multiskan MK3 microplate reader after addition of substrates. Wells with PBS-T only were used as negative controls.
Western blotting (WB)
The reactivity of produced rabbit anti-GST-VP1 hyperimmune serum was verified by WB. Briefly, culture medium collected from sf9 cells infected with recombinant baculovirus expressing JZ402 VP1 or wild-type baculovirus were boiled in loading buffer and loaded into SDS-PAGE gel. Separated proteins were transferred to polyvinylidene fluoride (PVDF) membrane. The PVDF membrane was blocked by PBS-T containing 1% BSA before the addition of rabbit anti-GST-VP1 hyperimmune serum diluted in PBS-T containing 1% BSA. The unbound immunoglobulin was removed by washing with PBS-T for three times. HRP-conjugated goat anti-rabbit IgG diluted in PBS-T was then added and the membrane was washed for another three more times before the addition of developing agents.
Indirect enzyme-linked immunosorbent assay (ELISA)
Overlapping peptide series (15-mer) covering the total P domain of GII.3 VP1 protein were synthesized and indirect ELISA was used to test the reactivity of these peptides with hyperimmune serum produced against JZ402 VP1-assembled VLPs and GST-VP1, respectively. Peptide series covering the S domain of GII.3 VP1 protein as reported previously were used to test their reactivity with hyperimmune serum against GST-VP1 [16]. Briefly, peptides diluted in PBS at 3 µg/ml were coated onto 96-well microplate and then the plate was blocked with PBS-T containing 1% BSA. Hyperimmune sera diluted in PBS containing 0.05% Tween-20 (PBS-T) at 1:1000 were added to above wells and the plate was incubated at 37 °C for 1 h. The plate was washed five times with PBS-T and then HRP-conjugated goat anti-rabbit or goat anti-guinea pig immunoglobulin was added. The plate was further incubated at 37 °C for 30 min and then developing agents were added. The signal was detected at 450 nm using microplate reader. GII.3 NoV VLPs coated wells were used as positive control and PBS-T added only wells were used as negative control.
Production of hyperimmune sera against peptide-BSA conjugates
The reactivity of peptides against rabbit anti-JZ402 VLP hyperimmune serum was determined by indirect ELISA as mentioned above. Serum-reactive peptides that were located in the P2 domain were conjugated with BSA for rabbit immunization. The immunization procedure was the same as described above and the amount of antigens for each immunization was 500 µg. The IgG titers of hyperimmune serum against each naked peptide were determined by indirect ELISA. Naked peptides were coated at 2 µg/ml. The titer was defined as the reciprocal value of serum dilutions that gave a value ≥ 2.1 folds of preimmunized serum value.
Salivary HBGAs-VLP binding blockade assay
In vitro salivary HBGAs-VLP binding blockade assay was performed basically the same as described previously [14]. HBGAs in saliva samples collected from individuals with blood type A, B, O, and AB were determined by using monoclonal antibodies against Lewis a, Lewis b, Lewis x, Lewis y, blood group A antigen, blood group B antigen, blood group H1 (O) antigen, and blood group H2 antigen (Abcam, UK). Briefly, saliva samples were boiled and coated onto plate in carbonate-bicarbonate buffer (pH 9.6) at 1:1000 or 1:100 and then the plate was blocked with PBS-T containing 1% BSA or 5% dried skimmed milk. ZZ3-34 NoV VLPs (2 µg) were predigested with trypsin at a final concentration of 200 µg/ml by incubating at 37 °C for 30 min [10]. JZ403 and trypsin digested and not digested ZZ3-34 NoV VLPs at 0.5 µg/ml were added to above wells with or without rabbit anti-JZ402 NoV VLPs, GST-VP1, JZ403 NoV VLPs, BSA-peptide conjugates, and guinea pig anti- GII.4-VP1/GII.3-P2 hyperimmune sera at 1:200. The plate was incubated at 37 °C for 1 h and then mixed rabbit anti-JZ402 and JZ403 NoV VLPs hyperimmune sera (volume ratio of 1:1) at 1:1000 were added. The plate was incubated at 37 °C for 1 h before the addition of HRP-conjugated goat anti-rabbit IgG. The plate was further incubated at 37 °C for 30 min and then substrates were added. The signal was detected at 450 nm using microplate reader. The plate was washed five times with PBS-T after each incubation step. Wells with PBS-T only were used as negative controls. The blocking index was calculated in % as (mean OD without sera-mean OD with sera)/mean OD without sera × 100%.
Results
Rabbit anti-GST-VP1 hyperimmune serum recognized GII.3 NoV VP1 proteins
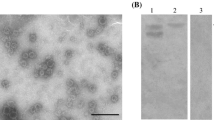
To express GST-VP1, positive clones containing correct insert were tested for protein expression. After sonication and centrifugation, supernatants and insoluble pellets from lyzed bacteria were analyzed by SDS-PAGE. Target bands with molecular weight of approximately 80 kDa (60 kDa for VP1 and 23 kD for GST) were detected in the pellet fraction, suggesting that GST-VP1 was expressed as inclusion bodies (data not shown). To simplify purification process, insoluble GST-VP1 was first separated by SDS-PAGE and then cut out from gel for recovery by electroelution method. Several grams of soluble GST-VP1 protein were produced in this way. Rabbit anti-GST-VP1 hyperimmune serum was produced by immunizing rabbit intradermally. Serum was collected after four immunizations and its specificity in recognizing GII.3 NoV VP1 proteins was determined by WB. As shown in Fig. 1, the produced rabbit anti-GST-VP1 hyperimmune serum specifically recognized expressed GII.3 VP1 using recombinant baculovirus expression system and no band was observed in the sample containing wild-type baculovirus.

Western blot. Supernatants collected from wild type (WT) and recombinant baculoviruses harboring the VP1 gene of GII.3 NoV (rBac-VP1) infected sf9 cells were loaded onto gels and separated proteins were detected by using rabbit anti-GST-VP1 fusion proteins hyperimmune serum as primary antibody
Produced hyperimmune sera exhibited high IgG titer against immunogens
The IgG titers of rabbit anti-GST-VP1 hyperimmune serum against GST-VP1, ZZ3-34, JZ403, and chimeric GII.4-VP1/GII.3-P2 NoV VLPs by indirect ELISA were 1,280,000, 20,000, 160,000, and 10,000, respectively. For Guinea pig anti-GII.4-VP1/GII.3-P2 hyperimmune serum, the specific IgG titer by using coated GII.4-VP1/GII.3-P2 antigen was 1,280,000 while it was 64,000 and 8000 for ZZ3-34 and JZ403 NoV VLPs, respectively.
Peptides exhibiting binding signals were primarily located in the P2 domain
Indirect ELISA was used to determine peptides that were reactive with produced hyperimmune sera. A total of 27 overlapping peptides (Table 1) covering the P domain sequence of GII.3 VP1 were tested for their reactivities against hyperimmune serum produced against JZ402 NoV VP1-assembled VLPs and GST-VP1, respectively. As was shown in Fig. 2a, peptide-1, 2, 3, 5, 6 ,7, 9, 13, 16, 22, and 27 exhibited binding signals and of which, peptide-5, 7, 9, 13, 16, and 27 exhibited stronger binding activities. Peptide-5, 6, 7, 9, 13, and 16 were located in the P2 domain and above results indicated that linear epitopes in JZ402 NoV VP1 were mainly located in the P2 subdomain. Surprisingly, none the 27 peptides derived from the P domain exhibited reactivities with hyperimmune serum produced against GST-VP1 (Fig. 2b). Binding signals were detected by using peptides derived from the S domain and the reactive peptides were located between the amino acids 31–60 and 93–117 (Table 2; Fig. 3).

Reactivity of peptides against rabbit anti-GII.3 NoV VLPs (a) and GST-VP1 (b) hyperimmune serum, respectively. Peptide sets covering the P2 domain of GII.3 NoV VP1 were coated onto microplate wells at 3 µg/ml and rabbit anti-GII.3 NoV VLPs or GST-VP1 hyperimmune serum diluted in PBS-T at 1:1000 was used. GII.3 NoV VLPs at 0.25 µg/ml was used as control. PBS-T was used as blank control

Reactivity of peptides against rabbit anti-GST-VP1 hyperimmune serum. Peptide sets covering the S domain of GII.3 NoV VP1 were coated onto microplate wells at 2.5 µg/ml and rabbit anti-GII.3 NoV VLPs hyperimmune serum diluted in PBS-T at 1:1000 was used. GII.3 NoV VLPs at 0.25 µg/ml was used as control. PBS-T was used as blank control
Hyperimmune serum against GII.4-VP1/GII.3-P2 exhibited blocking effects
In vitro salivary HBGAs-VLP binding blockade assay was used to characterize the ability of produced hyperimmune sera in blocking the binding of JZ403 NoV VLPs and trypsin digested ZZ3-34 VLPs against salivary HBGAs. As was shown in Fig. 4, rabbit anti-JZ402 and JZ403 NoV VLPs hyperimmune sera blocked the binding of ZZ3-34 and JZ403 NoV VLPs to salivary HBGAs of all blood types, respectively, with no or weak cross-blocking effects. Rabbit anti-GST-VP1 hyperimmune serum exhibited weak to moderate blocking effects against the binding of ZZ3-34 and JZ403 NoV VLPs to salivary HBGAs. Compared with ZZ3-34 VLPs, the blocking effects were more obvious for JZ403 VLPs, especially for blood type AB and O salivary HBGAs. Guinea pig anti-GII.4-VP1/GII.3-P2 exhibited no blocking effects against the binding of JZ403 VLPs to salivary HBGAs of all blood types while it blocked the binding of ZZ3-34 VLPs to salivary HBGAs of all blood types.

In vitro VLP-salivary HBGAs binding blockade assay. The blocking effects of rabbit anti-GST-VP1 (GST-VP1 PcAb), JZ402, and JZ403 NoV VLPs hyperimmune sera, as well as guinea pig anti- GII.4-VP1/GII.3-P2 VLPs (CP PcAb) hyperimmune serum against the binding of ZZ3-34 and JZ403 NoV VLPs to blood type A (a), B (b), AB (c), and O (d) salivary HBGAs were evaluated. ZZ3-34 and JZ403 NoV VLPs were used at 0.5 µg/ml and hyperimmune serum was used at 1:200. PcAb, polyclonal antibodies; CP PcAb, guinea pig anti- GII.4-VP1/GII.3-P2 VLPs hyperimmune serum. NC, negative control. Experiments were repeated at least three time and data were expressed as mean ± standard deviation
Hyperimmune serum against Peptide-BSA conjugates exhibited no blocking effects
As antibodies towards the P2 domain of GII.3 VP1 exhibited the ability of blocking the binding of cleaved GII.3 NoV VLPs against salivary HBGAs, it was thus supposed that the HBGAs blocking antibodies primarily bound the P2 domain. To test if antibodies binding linear epitopes in the P2 domain possess HBGAs blocking effects, peptide-5, 7, 9, and 13 were conjugated with BSA and used to immunize rabbits separately. Immunization of rabbits with the four peptide-BSA conjugates led to varied titers of peptide-specific IgG. The IgG titers of produced immune sera against peptide-5, 7, 9, and 13 were 128,000, 512,000, 512,000, and 128,000, respectively. The IgG titers of peptide-specific immune sera against GII.3 NoV VP1 proteins were 8000, 128,000, 8000, and 16,000, respectively. The reactivity of peptide-specific immune sera against GII.3 NoV VP1 proteins indicated that these peptides existed as linear epitopes in full-length VP1 proteins. To determine if antibodies binding linear epitopes in the P2 domain have HBGAs blocking effects, in vitro salivary HBGAs-VLP binding blockade assay was performed. As shown in Fig. 5, none of these peptide-specific immune sera showed HBGAs blocking effects, suggesting that HBGAs blocking antibodies might bind conformational epitopes.

In vitro VLP-salivary HBGAs binding blockade assay. The blocking effects of rabbit anti-P5, P7, P9, and P13 hyperimmune sera against the binding of ZZ3-34 to blood type A (a), B (b), AB (c), and O (d) salivary HBGAs were evaluated. ZZ3-34 was used at 0.5 µg/ml and hyperimmune serum was used at 1:200. PcAb, polyclonal antibodies; P5, peptide-5; P7, peptide-7; P9, peptide-9; P13, peptide-13. Rabbit anti-JZ402 hyperimmune serum at 1:200 was used as positive control. NC, negative control. Experiments were repeated at least three time and data were expressed as mean ± standard deviation
Discussion
Till now, there are no concrete data about the HBGAs binding epitopes for GII.3 NoV and all available data are derived from sequence alignment and structure simulation by comparing with that of GII.4 NoV capsid proteins. The difficulty in obtaining information about HBGAs binding epitopes for GII.3 NoV might be due to weak or absence of binding of GII.3 NoV VLPs to synthetic and salivary HBGAs. In our previous study, we successfully established an in vitro HBGAs binding blockade assay for GII.3 NoV VLPs by using trypsin digestion. We found that the binding of trypsin digested GII.3 NoV VLPs to salivary HBGAs cannot be blocked by heterotypic immune sera from the same or different genogroups [14]. In this study, we further characterized the binding regions and binding epitope types of HBGA blocking antibodies for GII.3 NoV VLPs by using immune sera produced against different antigens, including previously characterized chimeric antigen GII.4-VP1/GII.3-P2.
To determine that production of HBGAs blocking antibodies requires expressed VP1 proteins in correct conformation, we first expressed fusion protein GST-VP1, as which might not exhibit correct conformation even if it was expressed in soluble form. The expressed GST-VP1 protein existed as inclusion bodies and immunization of which was not supposed to induce the production of correct conformation-dependent antibodies. If the binding epitopes of the HBGA blocking antibody are conformation-dependent, then the immune serum against GST-VP1 should not have blocking effects. In vitro HBGA binding blockade assay confirmed this hypothesis. It should be noted that moderate blocking effects were also observed for blood type AB and O salivary samples. As the VP1 sequence in GST-VP1 was from JZ402 strain, the stronger blocking effects against JZ403 VLPs might be explained by two possibilities: first, solubilized GST-VP1 protein exhibited conformational epitopes deviating from GII.3 NoV VLPs but similar to those of JZ403 VLPs and antibodies against those epitopes blocked the binding of JZ403 VLPs to blood type AB and O salivary HBGAs. Second, there were linear epitopes located in the S domain or other sites except HBGAs binding sites, bound by corresponding antibodies to induce conformational changes, thus leading to ablation of HBGAs binding ability.
To locate the binding regions of the HBGA blocking antibodies, immune serum against previously characterized chimeric protein GII.4-VP1/GII.3-P2 was produced. Guinea pig anti-GII.4-VP1/GII.3-P2 serum exhibited no or very weak blocking effects against the binding of JZ403 NoV VLPs to salivary HBGAs while blocked the binding of trypsin digested ZZ3-34 NoV VLPs to salivary HBGAs, suggesting that HBGA binding blocking antibodies primarily targets the P2 domain.
To further characterize the epitope types (linear or conformational) that mediate binding of HBGAs blocking antibodies, we first screened for peptides covering the P2 domain that were reactive with anti-GII.3 NoV VLP hyperimmune sera, then conjugated those reactive peptides with BSA for immunization and finally performed in vitro HBGAs blocking assay by using produced peptide-specific immune sera. We hypothesize that the HBGAs blocking antibodies target the conformational epitopes, but not the linear epitopes, and antibodies targeting the linear epitopes in this region should exhibit no HBGAs blocking effects. Based on our results, peptide-5, 7, 9, and 13 exhibited binding activities against rabbit hyperimmune serum and immunization in rabbits using these peptides conjugated with BSA led to the production of peptide-specific antibodies. As these peptide-specific immune sera also recognized GII.3 NoV VP1 proteins, it was reasonable to think that antibodies targeting these peptides were similar with those that were induced by native proteins. In vitro HBGAs blocking assay indicated that none of these peptide-specific immune sera exhibited HBGAs blocking effects, suggesting monoclonal antibodies that bind linear epitopes might not have blocking effects.
Combining above data it is suggested that HBGA blocking antibodies against GII.3 NoV primarily bind the P2 region and most probably are conformation-dependent. The latter assumption requires further verification by using conformation-dependent monoclonal antibodies with blocking effects. Still more data are required in the future to confirm our conclusion as there are questions remain to be answered. For example, based on protein homology modeling, the surface exposed loop region in the VP1 of GII.3 NoVs might be involved in HBGA binding when compared with that of GII.4 NoV (epitope A [294, 296 to 298, 368 and 372]) and trypsin cleavage at this region might weaken or totally eliminate the binding capacity of this epitope to HBGAs [17, 18]. In fact, our recently conducted experiment indicated that ZZ3-34 NoV VLPs exhibited weak binding and loss of binding ability against synthetic LeX and LeY antigens before and after trypsin digestion, respectively. Thus, the observed blocking effects in this study might not actually reflect the complete binding epitopes. It has been reported that GII.3 strains have at least two binding specificities: one secretor-gene-dependent related to alpha1,2-fucosylated carbohydrates and another related to alpha2,3-sialylated carbohydrates of the type 2 chain [19]. Trypsin digestion of GII.3 NoV VLPs might affect its binding against alpha2,3-sialylated carbohydrates as LeX and LeY antigens have type 2 chains.
Based on our data, the GII.4-VP1/GII.3-P2 chimeric protein might be a good candidate antigen for the screening of HBGAs blocking antibodies during mAb production. mAbs exhibit binding activity against GII.4-VP1/GII.3-P2 but not JZ403 NoV VLPs and peptide arrays have a greater chance of being HBGA blocking antibody. In summary, the data and antigens provided in this study might be useful for the clarification of HBGAs binding epitopes for GII.3 NoVs.
References
Prasad B, Hardy M, Dokland T, Bella J, Rossmann M, Estes M (1999) Science 286:287–290
Prasad B, Rothnagel R, Jiang X, Estes M (1994) J Virol 68:5117–5125
Vinjé J (2015) J Clin Microbiol 53:373–381
Bu W, Mamedova A, Tan M, Xia M, Jiang X, Hegde RS (2008) J Virol 82:5340–5347
Cao S, Lou Z, Tan M, Chen Y, Liu Y et al (2007) J Virol 81:5949–5957
Choi J, Hutson A, Estes M, Prasad B (2008) Proc Natl Acad Sci USA 105:9175–9180
Chen Y, Tan M, Xia M, Hao N, Zhang X et al (2011) PLoS Pathog 7:e1002152
Lochridge V, Jutila K, Graff J, Hardy M (2005) J Gen Virol 86:2799–2806
Tan M, Huang P, Meller J, Zhong W, Farkas T, Jiang X (2003) J Virol 77:12562–12571
Tan M, Xia M, Cao S, Huang P, Farkas T et al (2008) Virology 379:324–334
Boon D, Mahar J, Abente E, Kirkwood C, Purcell R et al (2011) J Virol 85:8656–8666
Huo Y, Wan X, Ling T, Shen S (2016) Microb Pathog 90:78–83
K S-L ME, K A-B A, dR SA et al (2015) Clin Microbiol Infect 21:874.e1–874.e8
Huo Y, Wang W, Zheng L, Chen X, Shen S, Wang M (2017) Virus Res 240:18–24
Huo Y, Wang W, Ling T, Wan X, Ding L et al (2016) Virus Res 224:1–5
W W, Q JLXCSL W, et al (2018) J Med Virol 90:671–676
Donaldson E, Lindesmith L, Lobue A, Baric R (2008) Immunol Rev 225:190–211
Kumar S, Ochoa W, Kobayashi S, Reddy V (2007) J Virol 81:1119–1128
Rydell G, Nilsson J, Rodriguez-Diaz J, Ruvoën-Clouet N, Svensson L et al (2009) Glycobiology 19:309–320
Acknowledgements
We thank Qiong Fan, Department of Biochemistry, Institute of Basic Medical Sciences, University of Oslo and Xiaoran Li, Department of Pathology, University of Texas, MD Anderson Cancer Center for editing our manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
Animal experiments were performed in accordance with the guidelines of Chinese Council on Animal Care. The research protocol was approved by Animal Care and Use Committee of the Wuhan Institute of Biological Products (WIBP).
Additional information
Edited by Zhen F. Fu.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ma, S., Zhang, F., Zheng, L. et al. Linear epitope binding antibodies against GII.3 Norovirus exhibit no histo-blood group antigens (HBGAs) blocking effects. Virus Genes 55, 280–289 (2019). https://doi.org/10.1007/s11262-019-01644-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-019-01644-4