
Abstract
Three white spot syndrome virus (WSSV) isolates of different virulence were identified in our previous study, the high-virulent strain WSSV-CN01, the moderate-virulent strain WSSV-CN02 and the low-virulent strain WSSV-CN03. In this study, the genomes of these three WSSV isolates were sequenced, annotated and compared. The genome sizes for WSSV-CN01, WSSV-CN02, and WSSV-CN03 are 309,286, 294,261, and 284,148 bp, bearing 177, 164, and 154 putative protein-coding genes, respectively. The genomic variations including insertions, deletions, and substitutions were investigated. Thirty four genes show >20% variation in their sequences in WSSV-CN02 or WSSV-CN03, in comparison with WSSV-CN01, including six envelope protein genes (wsv237/vp41A, wsv238/vp52A, wsv338/vp62, wsv339/vp39, wsv077/vp36A, and wsv242/vp41B), and two immediate-early genes (wsv108 and wsv178). The genomic variations among WSSV isolates of different virulence, especially those in the coding regions, certainly provide new insight into the understanding of the molecular basis of WSSV pathogenesis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
White spot syndrome virus (WSSV) is a large enveloped double-stranded DNA virus that belongs to the family Nimaviridae, genus Whispovirus. It is elliptical in shape, about 80–120 nm in width and 250–380 nm in length, with a filamentous appendage at one end. WSSV infects many species of aquatic crustaceans including penaeid shrimp, crayfish, crab, and lobster, and it is the most devastating viral pathogen of aquacultured shrimp [1–3].
So far, the complete genome sequences have been determined for five WSSV isolates, including WSSV-CN strain (GenBank accession number AF332093) [4], WSSV-TH strain (GenBank accession number AF369029) [5], WSSV-TW strain (GenBank accession number AF440570) [6], WSSV-KR strain (GenBank accession number JX515788) [7], and WSSV-EG3 strain (GenBank accession number KR083866). It is notable that the sequence of WSSV-EG3 is almost identical to that of WSSV-CN. The WSSV genomes range from 293 to 307 kb, predicted to encode ~180 proteins.
Although the overall nucleotide identity among WSSV isolates is very high, two major variable regions have been found in the genomes of WSSV isolates, including a major deletion region at ORF23/24 of the WSSV-TH genome (corresponding to wsv477/502 in WSSV-CN), and a major variable region at ORF14/15 (corresponding to wsv461/464 in WSSV-CN) [8]. Sequence alignments have also shown difference in the variable number tandem repeats (VNTRs) located in ORF75 (wsv129), ORF94 (wsv178), and ORF125 (wsv249) [8, 9]. These variable DNA loci are currently used as genetic marker to identify WSSV variants and analyze the patterns of viral spreading [8–12].
Although WSSV isolates obtained from different geographical regions are similar in morphology and host range, variations in virulence have been widely observed [9, 13–17]. The relationship between genetic variations in the variable regions (ORF14/15, ORF23/24 and VNTRs) and the change in virulence has been investigated in several studies, but the results are conflicting. In 2005, Marks et al. [9] compared the virulence of two WSSV isolates by analyzing shrimp mortality rate and competitive fitness, and showed that WSSV-TH was more virulent than TH-96-II. Because TH-96-II has a relatively larger genome, they suggested that smaller genome size might give to WSSV both a replication advantage and an increase in fitness. On the contrary, Lan et al. [17] showed that a 4.8 kb deletion of the WSSV genome led to a dramatic decrease in virulence. In addition, studies focusing on the VNTRs suggested that WSSV isolates with fewer repeat units were more virulent [14, 18, 19]. So far, there is no enough evidence to support a link between genome size and replicative fitness or virulence for WSSV. Moreover, in all the previous studies concerning the virulence-associated genomic variations in WSSV genomes, only the highly variable regions were investigated. No comparison has been made at the full-length genome level.
In our previous study, three WSSV isolates significantly different in virulence ranking from high (WSSV-CN01), moderate (WSSV-CN020) to low (WSSV-CN03) were identified. These WSSV isolates have similar replication kinetics in the host, but they can induce distinct immune responses, and the median lethal times of the animals infected by these isolates are significantly different [13]. Therefore, the isolate with higher virulence may encode virulent factors that lead to more severe disease in the hosts.
To understand the molecular basis contributing to the variation of WSSV pathogenicity, the genome of WSSV-CN01, WSSV-CN02, and WSSV-CN03 were sequenced and compared in this study. Genomic variations potentially associated with the virulence were identified. This is the first attempt to identify virulence-associated genomic variation of WSSV by comparing the complete genomes of isolates of different virulence, which provides important information for the understanding of WSSV pathogenesis.
Materials and methods
Virus strains and purification of virions
The virus strain WSSV-CN01 was obtained from infected Marsupenaeus japonicus from Xiamen, China in 1994. WSSV-CN02 strain was isolated from infected crayfish Procambarus clarkii from Xiamen, China in 2010. WSSV-CN03 strain was isolated from infected Litopenaeus vannamei from Zhangpu, China in 2010 [13]. Virions were propagated in P. clarkii and purified as previously described [20]. The concentration of purified virions was determined using the method established by Zhou et al. [21].
Genome sequencing and analyses
WSSV genomic DNA was prepared from purified virions as described previously [22]. Viral genomes were sequenced using 454 sequencing technology and assembled using the GS de novo assembler software (Version 2.8) by Shanghai Majorbio Bio-pharm Biotechnology Co., Ltd. The genomes were annotated and analyzed using Geneious 10.0.5. The open reading frames (ORFs) of 60 aa or larger with minimum overlap were identified as potential protein-coding genes. The genome structure was analyzed using the “align whole genomes” function of MAUVE [23]. The identities between the genomic sequences of WSSV-CN and the other six fully sequenced WSSV isolates were determined by the pairwise alignment of Geneious.
Phylogenetic analysis
To infer the evolutionary relationships among seven fully sequenced WSSV isolates, multiple sequence alignments of the whole genome were performed with ClustalX [24]. A maximum-likelihood phylogenetic tree was constructed using PhyML with Smart Model Selection (http://www.atgc-montpellier.fr/phyml-sms/) [25]. The TN93 + G + F model [26] was automatically selected by PhyML as the best model to infer the phylogenetic tree. Bootstrap tests were carried out with 1000 replicates. FigTree (version 1.4.3) (http://tree.bio.ed.ac.uk/software/figtree/) was employed for tree visualization.
Analysis of the large-scale variations in the WSSV genomes by PCR
To verify the sequencing results, genomic DNA was purified from WSSV isolates as described above. The regions with large-scale insertion or deletion among different isolates were amplified by PCR, and analyzed by electrophoresis. Moreover, the gene splitting and merging events were confirmed by PCR and sequencing of the regions. The primers used to analyze large-scale genomic variations (GV1-6) were listed in Supplementary Table 1.
Reverse transcription PCR (RT-PCR) analysis
Crayfish were injected with WSSV virions (1 × 104/individual). Total RNA was extracted from the muscle tissue at 48 h post-infection using TRI REAGENT (Molecular Research Center, Inc.) following the manufacturer’s instructions. After RNasefree DNase I (Takara) treatment to remove residual DNA, the first-strand of cDNA was synthesized from total RNA with oligo(dT)18-primer by reverse transcriptase (Roche Diagnostic). Genes of interest were detected by PCR (denaturing at 94 °C for 5 min; 30 cycles at 94 °C for 1 min, 58 °C for 30 s, and 72 °C for 30 s; final extension at 72 °C for 5 min) with primers listed in Supplementary Table 2, and the PCR products were analyzed by electrophoresis.
Antibodies and Western blotting
Polyclonal antibodies against WSSV structural proteins were produced either in mouse (for VP41A and VP28) or in rabbit (for VP62, VP52A, and VP39) by Shanghai Immune Biotech Ltd, China. Purified virions were lysed with SDS-loading buffer and viral structural proteins were resolved by SDS-PAGE, and transferred to polyvinylidene fluoride (PVDF) membranes (Immobilon-P, Millipore) by semi-dry electrotransfer. The membrane was blocked in Chemiluminescent Blocker (Millipore) and probed with specific primary antibodies and alkaline phosphatase-conjugated secondary antibody (Promega). The signals were visualized with 5-Bromo-4-chloro-3-indolyl phosphate/nitroblue tetrazolium.
Results
Genomic sequences of WSSV-CN01, WSSV-CN02, and WSSV-CN03
The complete nucleotide sequences of WSSV-CN01, WSSV-CN02, and WSSV-CN03 were determined, and deposited in the GenBank (accession numbers KT995472, KT995470, and KT995471, respectively). The genome of WSSV-CN01, WSSV-CN02, and WSSV-CN03 were assembled into contigs of 309,286, 294,261, and 284,148 bp, and the GC contents of the three WSSV strains are 40.9, 41.0, and 41.0%, respectively. The main characteristics of seven fully sequenced WSSV isolates are summarized in Table 1. The genomic sequences identities between the type strain WSSV-CN and the other six WSSV isolates were determined by pairwise alignment. Because the genomic sequence of WSSV-EG3 is almost identical to that of WSSV-CN, it is not listed in this table.
The schematic diagrams showing the organization of the circular genomes of WSSV-CN01, WSSV-CN02, and WSSV-CN03 are provided in Supplementary Fig. 1–3. The genomic sequences of WSSV-CN01, WSSV-CN02, and WSSV-CN03 were annotated and the ORFs were predicted. The nomenclature of ORFs was based on that of WSSV-CN [4]. The ORFs of 60 aa or larger with minimum overlap were identified as potential protein-coding genes. WSSV-CN01, WSSV-CN02, and WSSV-CN03 were predicted to encode 177, 164, and 154 hypothetical proteins, respectively. The location, orientation, size, and function of each predicted protein-coding gene are summarized in Supplementary Table 3.
Phylogenetic analysis of WSSV isolates
A maximum-likelihood phylogenetic tree based on whole genome shows the clustering pattern of 7 fully sequenced WSSV isolates (Fig. 1). WSSV-CN is closer to WSSV-KR, while WSSV-CN01 and WSSV-CN02 group with WSSV-TW. WSSV-CN03 and WSSV-TH are more distantly related to other WSSV isolates analyzed in this study. This tree is supported by relatively high bootstrap value.

Phylogenetic analysis of WSSV isolates. A maximum-likelihood tree was constructed based on the genomic sequences of seven fully sequenced WSSV isolates with 1000 bootstrap replicates (numbers = bootstrap percentage values)
Genomic variations of WSSV-CN01, WSSV-CN02, and WSSV-CN03
Variation in virulence was found among WSSV-CN01, WSSV-CN02, and WSSV-CN03 in our previous study [13]. We speculated that the high-virulent WSSV-CN01 might encode some virulence-associated factors that were absent or had a loss-of-function in the moderate-virulent WSSV-CN02 or low-virulent WSSV-CN03. To investigate the potential virulence-associated genomic variations, the genome sequences of WSSV-CN01, WSSV-CN02, and WSSV-CN03 were compared with that of the type strain WSSV-CN.
Although the genomic sequences and structures are highly conserved among the WSSV strains, there are six large-scale insertions or deletions, corresponding to locations 37421–38639 (deletion), 63379–64319 (deletion), 97491–99012 (deletion), 132914–138225 (deletion), 197938–200097 (deletion), and 272082 (insertion) in the genome of WSSV-CN (Supplementary Fig. 4). A few SNP and indels were also identified (Supplementary Table 4). It is notable that the large majority of the genomic variations are present in the protein-coding regions. The variations in the protein-coding regions, including insertions, deletions, and substitutions were investigated and summarized in Supplementary Table 3. The regions with large-scale insertion or deletion were further verified by PCR (Fig. 2). The gene splitting and merging events were confirmed by PCR amplification and sequencing (data not shown).

Analysis of the large-scale variations in WSSV genome by PCR. WSSV genomic DNA was purified from different isolates. The regions with large-scale insertion or deletion among different isolates (GV1-6) were amplified by PCR, and analyzed by electrophoresis. M, 100 bp DNA ladder, or Lambda DNA HindIII/EcoRI marker
Sequence alignment indicates that the polymorphology of WSSV-CN01, WSSV-CN02, and WSSV-CN03 genomic sequences is mainly occurred in the two well-known variable regions of WSSV, which locate at wsv461/464 (Fig. 3a) and wsv477/502 (Fig. 3b) in WSSV-CN genome (corresponding to ORF14/15 and ORF23/24 in WSSV-TH) [8]. Compared with the type strain WSSV-CN, WSSV-CN01 genome carries an insertion of ~5 kb at the wsv461/464 (corresponding to ORF14/15 in WSSV-TH) variable site. This segment was not found in the genome of any other WSSV isolates except TH-96-II (GenBank accession No. AY753327) (Fig. 3a) [9]. Four genes named wsv463a, wsv463b, wsv463c, and wsv463d were identified in this region, which corresponded to ORF I + II, III, IV, V of WSSV TH-96-II [9]. Moreover, in WSSV-CN02, wsv461 and wsv463 are merged into one ORF, which corresponded to WSSV521 of WSSV-TW strain. In WSSV-CN03, wsv463 is completely absent while wsv461 and wsv464 contain large deletions.

Schematic representation of the variable regions wsv461/464 (ORF14/15) and wsv477/502 (ORF23/24) of WSSV isolates. The variable regions wsv461/464 (ORF14/15) (a) and wsv477/502 (ORF23/24) (b) in the genomes of WSSV isolates CN, CN01, CN02, CN03, KR, TH-96-II, TH, and TW are compared. c Transposition of wsv486 in WSSV-CN03. The positions of the DNA fragments are indicated with numbers above each isolate. These position numbers are consistent with the position numbers of the genomic sequence deposited in GenBank for each isolate. The positions, lengths, and transcriptional directions of the protein-coding genes are indicated with arrows. Deletions are indicated with dot lines. The names of ORFs are shown on the top of each figure using WSSV-CN naming system. The ORFs that are not present in WSSV-CN or the ORFs of strains that use a different naming system are indicated separately
The variation region wsv477/502 in WSSV-CN01 (corresponding to ORF23/24 in WSSV-TH) is similar to that of WSSV-TW strain. In comparison with WSSV-CN, both WSSV-CN01 and WSSV-TW contain a ~1.1 kb insertion in wsv495 which greatly enlarges the ORF. Wsv489–wsv497 are absent in WSSV-CN02, while wsv482, wsv484, and wsv489–wsv497 are absent in WSSV-CN03. The comparison of this variable region of different WSSV isolates is shown in Fig. 3b. In addition, the DNA fragment containing wsv486 is translocated to 104836–102527 in WSSV-CN03 and replaces wsv195 (Fig. 3c). The up-stream and down-stream sequences of the fragment are inverted repeats, which is a typical characteristic of transposable elements [27]. Since this fragment does not encode a transposase, it may be a non-autonomous transposable element. But we cannot exclude the possibility that the transposase has been lost during evolution. This translocation has not been observed in other WSSV isolates.
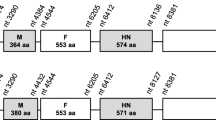
Apart from the two variable regions mentioned above, complete deletion of genes was found at wsv231/242. Compared with WSSV-CN, WSSV-CN02 has a ~ 4 kb deletion in this region, while WSSV-CN03 has two deletions of ~3.3 kb and ~0.4 kb deletions. Wsv234 and wsv237 are absent in WSSV-CN02, while wsv231 and wsv238 bear large-scale deletions. In WSSV-CN03, wsv237 and wsv238 are absent and wsv242 contain a 153 aa deletion (Fig. 4a). Moreover, wsv338 and 339 were not found in the genome of WSSV-CN03 (Fig. 4b). It is notable that wsv237, wsv238, wsv242, wsv338, and wsv339 are WSSV envelop protein genes, encoding VP41A, VP52A, VP41B, VP62, and VP39. These regions are conserved in WSSV-CN, WSSV-TH, WSSV-TW, and WSSV-KR.

Schematic representation of the variable regions wsv231/242 and wsv338/339. The variable regions, wsv231/242 and wsv338/339, which contain viral envelope protein genes are compared for WSSV-CN, -CN01, CN02, -CN03. The positions of the DNA fragments are indicated with numbers above each isolate. These position numbers are consistent with the genomic sequence deposited in GenBank for each isolate. The positions, lengths, and transcriptional directions of the protein-coding genes are indicated by arrows. Deletions are indicated with dot lines. The protein-coding genes are named in accordance with previous studies [4]
Three genes each with a VNTR have been identified as highly variable regions in WSSV genome as well, which are wsv129 (ORF75), wsv178 (ORF94), and wsv249 (ORF125) [8]. Wsv129 (ORF75) and wsv178 (ORF94) are completely absent in WSSV-CN03. The repeat units of wsv249 (ORF125) in WSSV-CN03 and WSSV-CN01 are both one unit shorter than that in WSSV-CN. In the case of WSSV-CN02, the number of repeat units reduces in wsv129 and wsv249 (Supplementary Table 3).
Splitting of ORFs was noticed in these isolates as well. The immediate-early (IE) gene wsv108 splits into two ORFs, wsv108a and wsv108b in WSSV-CN01, while envelope protein gene wsv077 (VP36A) splits into two ORFs (wsv077a and wsv077b) in WSSV-CN02 (Supplementary Table 3).
In addition to the large-scale variations mentioned above, there are also some small-scale variations present in WSSV-CN01, WSSV-CN02, and WSSV-CN03 in comparison with WSSV-CN, which also alter the aa sequence of viral genes (Supplementary Table 3).
We assume that genes containing >20% variation in their aa sequences, or completely absent in the less virulent strains are likely associated with the virulence. In comparison with the high-virulent strain WSSV-CN01, 30 genes contain >20% aa variation in WSSV-CN03 and 21 genes contain >20% aa variation in WSSV-CN02. Among them, 17 genes vary in both WSSV-CN02 and WSSV-CN03, while 13 genes vary exclusively in WSSV-CN03 and 4 genes vary exclusively in WSSV-CN02. Some of these genes may be associated with WSSV virulence (Table 2).
It is notable that two types of genes are conserved among different isolates (Supplementary Table 3): (1) Genes involved in DNA replication and nucleotide metabolism, including thymidylate synthase (wsv067), dUTPase (wsv112), ribonucleotide reductase 1 (rr1, wsv172), ribonucleotide reductase 2 (rr2, wsv188), nuclease (wsv191), thymidine kinase-thymidylate kinase (TK-TMK, wsv395), and DNA polymerase (wsv514); (2) Major structure protein genes, including the four major envelop protein VP28 (wsv421), VP26 (wsv311), VP24 (wsv002), VP19 (wsv414), the major capsid protein VP664 (wsv360), and the DNA binding protein VP15 (wsv214) [28–30].
Analysis of the expression of genes varied among WSSV isolates
RT-PCR was performed to verify the transcription of 20 genes which were found to be absent in WSSV-CN02 or WSSV-CN03 by genomic sequencing. Total mRNA was extracted from WSSV-infected crayfish at 48 h post-infection, and the transcription of candidate genes was analyzed. Because the viral IE gene ie1 (wsv069) and major envelope protein gene vp28 (wsv421) are present in all WSSV strains, they were chosen as positive controls, and the host actin gene were used as a loading control. As shown in Fig. 5, ie1 and vp28 were transcribed in all three WSSV strains, and all the candidate genes were transcribed in WSSV-CN01-infected crayfish. Eighteen candidate genes (wsv073, wsv178, wsv237, wsv238, wsv338, wsv339, wsv463a, wsv463b, wsv463c, wsv463d, wsv482, wsv484, wsv489, wsv490, wsv492, wsv493, wsv495, wsv497) were not transcribed in WSSV-CN03-infected crayfish, while 13 candidate genes (wsv204, wsv234, wsv237, wsv463a, wsv463b, wsv463c, wsv463d, wsv489, wsv490, wsv492, wsv493, wsv495, wsv497) were not transcribed in WSSV-CN02-infected crayfish. These data are consistent with the sequencing results.

Transcriptional analysis of protein-coding genes varied in WSSV-CN, WSSV-CN01, WSSV-CN02, and WSSV-CN03. Total RNAs were extracted from the muscles of crayfish infected with different WSSV isolates at 48 h post-infection. The transcription of target genes was analyzed by RT-PCR. Actin gene was used as a loading control. The immediate-early gene ie1 and late gene vp28 were used as positive controls
According to the genomic sequencing results, there are four envelope genes absent in WSSV-CN02 or WSSV-CN03, viz. wsv237/vp41a, wsv238/vp52a, wsv338/vp62, and wsv339/vp39. To further confirm the result, the virions of WSSV-CN01, WSSV-CN02, and WSSV-CN03 were purified and the presence of these four envelope proteins was analyzed by Western blotting. The major envelope protein VP28 was used as a positive control. As shown in Fig. 6, VP28 was present in all the isolates. VP62, VP52A, VP41A, and VP39 were absent in WSSV-CN03 virions, while VP41A was absent in WSSV-CN02 virions. Although wsv238/vp52a was transcribed in WSSV-CN02-infected crayfish, we failed to detect VP52A in WSSV-CN02 virions. This might result from the 287 aa deletion in the N-terminus of VP52A in this isolate (Supplementary Table 3). Moreover, envelope protein gene wsv077/vp36a split into two ORFs in WSSV-CN02, and wsv242/vp41b is truncated in WSSV-CN03 (Supplementary Table 3). Due to the lack of the antibodies for VP36A and VP41B, the variations of these two envelope proteins were not characterized in this study.

Western blotting analysis of envelope proteins varied in WSSV-CN, WSSV-CN01, WSSV-CN02, and WSSV-CN03. Purified virions of WSSV-CN, WSSV-CN01, WSSV-CN02, and WSSV-CN03 were lysed in SDS-loading buffer, and the structural proteins were separated by SDS-PAGE and transferred to PVDF membranes. The membranes were probed with anti-VP62, anti-VP52A, anti-VP41A, anti-VP39, and anti-VP28 primary antibodies
Discussion
Three white spot syndrome virus (WSSV) isolates of different virulence were identified in our previous study, the high-virulent strain WSSV-CN01, the moderate-virulent strain WSSV-CN02, and the low-virulent strain WSSV-CN03 [13]. To understand the molecular basis for the difference in virulence, the complete genomic sequences of these three virulence-different WSSV isolates were sequenced and compared. The results show that WSSV-CN01 contains the largest genome among the three isolates investigated in this study. The genome of WSSV-CN01 is 15 kb larger than that of WSSV-CN02 and 25 kb larger than that of WSSV-CN03 (Table 1). The replication kinetics for WSSV-CN01, WSSV-CN02, and WSSV-CN03 is similar [13]. Therefore, there is not an obvious link between WSSV genome size and virulence. Instead, the isolates with higher virulence may encode virulent-associated factors that are absent or has a loss-of-function in less virulent isolates. Our findings are in contrast to some previous reports which proposed that genome shrinkage of WSSV was an adaptive process that might give the virus a replication advantage, and contributed to increased viral fitness and virulence [9, 14, 31].
To reveal the molecular basis of virulent difference, the genomic variations among WSSV-CN01, WSSV-CN02, and WSSV-CN03 were investigated, and the large majority of variations were found in the coding regions (Supplementary Table 3). In total, 34 proteins were found to contain >20% variation in the aa sequences among these three WSSV isolates (Table 2). We assume that some of them may be associated with WSSV virulence determination. Because the replication kinetic of WSSV-CN01, WSSV-CN02, and WSSV-CN03 are similar [13], these variable genes may not be essential for viral replication. Notably, four of the genes, wsv463a, wsv463b, wsv463c, and wsv463d, are also found in WSSV TH-96II which has been identified as a low-virulent isolate [9]. Although the virulence of TH-96II, WSSV-CN01, WSSV-CN02, and WSSV-CN03 has not been compared directly, we speculate that these four genes may play a minor role in virulence determination. Besides, we cannot exclude the possibility that the proteins with minor alternations in their aa sequence (≤20%) and the genomic variations in non-coding regions may also contribute to difference.
In comparison with WSSV-CN01, four envelope proteins (VP62, VP39, VP41A, and VP52A) are absent in WSSV-CN03, one envelope protein VP41B is truncated, and the envelope protein VP36A splits into two ORFs in WSSV-CN02 (Table 2, Supplementary Table 3; Fig. 6). Viral envelope proteins are important for maintaining the structure of the virions. However, no obvious morphological difference was observed for the WSSV isolates investigated in this study (data not shown). Compared with major envelope proteins VP24, VP26, VP28, and VP19, the amounts of VP36A, VP62, VP39, VP41A, VP52A, and VP41B in the virions are very low [28, 29, 32]. Therefore, the variations of these six proteins may have little effect on viral morphology. Moreover, viral envelope proteins may participate in viral invasion and signal transduction during viral infection. Research of other viruses showed that mutation in envelope protein may change viral virulence by interfering with the entry process or modulating host cells [33–35]. Because the entry process of WSSV-CN01, WSSV-CN02, and WSSV-CN03 has not been compared yet, whether alternation of the six envelope proteins affects WSSV entry remains to be explored. Interestingly, WSSV VP41B has been found to activate the expression of shrimp caspase gene by binding to its promoter [36]. Thus, the mutation of VP41B might affect its function in transcriptional regulation and in-turn modulate host immune responses [37].
In addition, variations were found in two IE genes, wsv108 and wsv178 (Table 2; Supplementary Table 3) [38]. Virus IE genes often encode regulatory proteins that control the expression of viral genes, alter the functions of host genes, or eliminate host immune defense [39, 40]. Although the functions of wsv108 and wsv178 are currently unknown, the deletion of these genes in the genome of low-virulent isolate suggests that they may contribute to WSSV pathogenesis.
The three WSSV VNTR regions are the compound 45 and 102-bp repeat units regions in ORF75 (wsv129), the 54-bp repeat units region in ORF94 (wsv178), and the 69-bp repeat units region in ORF125 (wsv249) [8]. Epidemiology researches suggested that the presence of less repeat units in VNTRs was significantly correlated with disease outbreaks/virulence [18]. However, in our study, ORF75 (wsv129) and ORF94 (wsv178) were completely absent in the genome of low-virulent isolate WSSV-CN03, and the repeat unit number of ORF125 (wsv249) is similar to that of WSSV-CN01. In the case of WSSV-CN02, reduction of repeat unit numbers of ORF75 (wsv129) and ORF125 (wsv249) was observed when compared with WSSV-CN01 (Supplementary Table 3). Therefore, whether there is a link between the number of repeat units in VNTR and WSSV virulence/fitness remains a question.
Conclusions
By analyzing genomic variations among WSSV isolates of different virulence, especially those in the coding regions, we identify 34 candidate genes that might be associated with WSSV virulence, which provide important information for the understanding of WSSV pathogenesis. Further experiments are necessary to investigate the function of these genes during viral infection and explore the mechanism of virulence determination.
References
A. Sanchez-Paz, Vet. Res. 41(6), 43 (2010)
J.H. Leu, F. Yang, X. Zhang, X. Xu, G.H. Kou, C.F. Lo, Curr. Top. Microbiol. Immunol. 328, 197–227 (2009)
C.M. Escobedo-Bonilla, V. Alday-Sanz, M. Wille, P. Sorgeloos, M.B. Pensaert, H.J. Nauwynck, J. Fish Dis. 31(1), 1–18 (2008)
F. Yang, J. He, X. Lin, Q. Li, D. Pan, X. Zhang, X. Xu, J. Virol. 75(23), 11811–11820 (2001)
M.C.W. van Hulten, J. Witteveldt, S. Peters, N. Kloosterboer, R. Tarchini, M. Fiers, H. Sandbrink, R.K. Lankhorst, J.M. Vlak, Virology 286(1), 7–22 (2001)
L.-L. Chen, H.-C. Wang, C.-J. Huang, S.-E. Peng, Y.-G. Chen, S.-J. Lin, W.-Y. Chen, C.-F. Dai, H.-T. Yu, C.-H. Wang, C.-F. Lo, G.-H. Kou, Virology 301(1), 136–147 (2002)
C.Y. Chai, J. Yoon, Y.S. Lee, Y.B. Kim, T.J. Choi, J. Microbiol. 51(5), 695–699 (2013)
H. Marks, R.W. Goldbach, J.M. Vlak, M.C. van Hulten, Arch. Virol. 149(4), 673–697 (2004)
H. Marks, J.J.A. van Duijse, D. Zuidema, M.C.W. van Hulten, J.M. Vlak, Virus Res. 110(1–2), 9–20 (2005)
B.T. Dieu, H. Marks, M.P. Zwart, J.M. Vlak, J. Gen. Virol. 91(Pt 5), 1164–1172 (2010)
M. de Jesus Duran-Avelar, R. Perez-Enriquez, J.F. Zambrano-Zaragoza, L. Montoya-Rodriguez, R. Vazquez-Juarez, N. Vibanco-Perez, Dis. Aquat. Organ. 114(1), 11–20 (2015)
G. Sablok, A. Sanchez-Paz, X. Wu, J. Ranjan, J. Kuo, I. Bulla, Arch. Virol. 157(12), 2357–2362 (2012)
M. Gao, F. Li, L. Xu, X. Zhu, Fish Shellfish Immunol. 39(1), 17–23 (2014)
B. Pradeep, I. Karunasagar, I. Karunasagar, J. Fish Dis. 32(9), 801–805 (2009)
S.E. Laramore, J. Scarpa, C.R. Laramore, J. Lin, J. Aquat. Anim. Health 21(2), 82–90 (2009)
M.M. Rahman, M. Corteel, C.M. Escobedo-Bonilla, M. Wille, V. Alday-Sanz, M.B. Pensaert, P. Sorgeloos, H.J. Nauwynck, Dis. Aquat. Organ. 79(3), 191–198 (2008)
Y. Lan, W. Lu, X. Xu, Virus Res. 90(1–2), 269–274 (2002)
T.T. Hoa, M.P. Zwart, N.T. Phuong, M.C. de Jong, J.M. Vlak, J. Fish Dis. 35(11), 817–826 (2012)
G. Waikhom, K.R. John, M.R. George, M.J.P. Jeyaseelan, Aquaculture 261(1), 54–63 (2006)
X. Xie, H. Li, L. Xu, F. Yang, Virus Res. 108(1–2), 63–67 (2005)
Q. Zhou, Y.P. Qi, F. Yang, J. Virol. Methods 146(1–2), 288–292 (2007)
F. Yang, W. Wang, R.Z. Chen, X. Xu, J. Virol. Methods 67(1), 1–4 (1997)
A.C. Darling, B. Mau, F.R. Blattner, N.T. Perna, Genome Res. 14(7), 1394–1403 (2004)
J. D. Thompson, T. J. Gibson, D. G. Higgins, Curr Protoc Bioinformatics. Chapter 2, Unit 2 3(2002)
S. Guindon, O. Gascuel, Syst. Biol. 52(5), 696–704 (2003)
K. Tamura, M. Nei, Mol. Biol. Evol. 10(3), 512–526 (1993)
C. Feschotte, N. Jiang, S.R. Wessler, Nat. Rev. Genet. 3(5), 329–341 (2002)
X. Xie, L. Xu, F. Yang, J. Virol. 80(21), 10615–10623 (2006)
J.M. Tsai, H.C. Wang, J.H. Leu, A.H. Wang, Y. Zhuang, P.J. Walker, G.H. Kou, C.F. Lo, J. Virol. 80(6), 3021–3029 (2006)
Y. Liu, J. Wu, H. Chen, C.L. Hew, J. Yan, Virology 408(2), 197–203 (2010)
M.P. Zwart, B.T. Dieu, L. Hemerik, J.M. Vlak, PLoS ONE 5(10), e13400 (2010)
Z. Li, Q. Lin, J. Chen, J.L. Wu, T.K. Lim, S.S. Loh, X. Tang, C.L. Hew, Mol. Cell. Proteom. 6(9), 1609–1620 (2007)
E. Baranowski, C.M. Ruiz-Jarabo, E. Domingo, Science 292(5519), 1102–1105 (2001)
L. de Borba, D.M. Strottmann, L. de Noronha, P.W. Mason, C.N. Dos Santos, PLoS Negl. Trop. Dis. 6(4), e1624 (2012)
G. Schlecht-Louf, M. Renard, M. Mangeney, C. Letzelter, A. Richaud, B. Ducos, I. Bouallaga, T. Heidmann, Proc. Natl. Acad. Sci. USA 107(8), 3782–3787 (2010)
H. Zuo, C. Chen, Y. Gao, J. Lin, C. Jin, W. Wang, Fish Shellfish Immunol. 30, 1188–1191 (2011)
L. Wang, B. Zhi, W.L. Wu, X.B. Zhang, Dev. Comp. Immunol. 32(6), 706–715 (2008)
F. Li, M. Li, W. Ke, Y. Ji, X. Bian, X. Yan, Virology 385(1), 267–274 (2009)
T.E. Morrison, A. Mauser, A. Klingelhutz, S.C. Kenney, J. Virol. 78(1), 544–549 (2004)
Y. Yuan, Methods Mol. Biol. 292, 231–244 (2005)
Acknowledgements
We thank Dr. Xiaofeng Shi for assistance in phylogenetic analysis, Dr. Mingliang Chen and Zhenping Zhong for proof-reading of the manuscript. This work was supported by the National Basic Research Program of China (973 Program, Grant Number 2012CB114401), the Natural Science Foundation of China (Grant Numbers 41376173 and 41276176), the China Agriculture Research System (Grant Number CARS-47), and the Fujian Marine Hi-tech Industry Program (Grant Number 2014-05).
Author contributions
FL and FY conceived and designed the experiments; MG performed the RT-PCR and Western blotting experiment; FL, MG, and FY participated in analyzing and interpretation of the genomic sequencing data; LX performed the virus purification, DNA extraction experiment, and PCR confirmation of the genomic variation. FL, MG, and FY drafted the manuscript. All authors read and approved the final manuscript. FL and MG contributed equally to this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
This research did not involve Human Participants or Animals.
Informed consent
This research did not involve Human Participants therefore no informed consent is required.
Additional information
Edited by A. Lorena Passarelli.
Fang Li and Meiling Gao have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig. 1
Schematic diagram showing the organization of the circular genome of WSSV-CN01. Schematic diagram showing the organization of the circular genome of WSSV-CN01. Positions of the predicted protein coding genes and their transcription directions are indicated with arrows (PDF 705 kb)
Supplementary Fig. 2
Schematic diagram showing the organization of the circular genome of WSSV-CN02. Schematic diagram showing the organization of the circular genome of WSSV-CN02. Positions of the predicted protein coding genes and their transcription directions are indicated with arrows (PDF 666 kb)
Supplementary Fig. 3
Schematic diagram showing the organization of the circular genome of WSSV-CN03. Schematic diagram showing the organization of the circular genome of WSSV-CN03. Positions of the predicted protein coding genes and their transcription directions are indicated with arrows (PDF 650 kb)
Supplementary Fig. 4
The graphic result of the whole-genome alignment of WSSV-CN, -CN02, CN02, and CN03. The genomic sequences of WSSV-CN, -CN02, CN02, and CN03 were aligned with the “align whole genomes” function of MAUVE. Homologous blocks are shown as identically colored regions. ORFs transcribed forward (above) and reverse (below) are indicated (PDF 702 kb)
Supplementary Table 1
Primers used to analyze large-scaled genomic variations of different WSSV strains (PDF 12 kb)
Supplementary Table 2
Primers used for RT-PCR analysis (PDF 13 kb)
Supplementary Table 3
Comparison of protein coding genes of WSSV-CN, -CN01, -CN02 and -CN03 (PDF 168 kb)
Supplementary Table 4
The number of indels and SNPs in the genomes of WSSV-CN01, -CN02 and –CN03 in comparison with WSSV-CN (PDF 10 kb)
Rights and permissions
About this article

Cite this article
Li, F., Gao, M., Xu, L. et al. Comparative genomic analysis of three white spot syndrome virus isolates of different virulence. Virus Genes 53, 249–258 (2017). https://doi.org/10.1007/s11262-016-1421-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-016-1421-z