
Abstract
Since the first outbreak of highly pathogenic avian influenza virus (HPAIV) subtype H5N1 in Bangladesh in 2007, the virus has been circulating among domestic poultry causing severe economic losses. To investigate the presence of HPAIV H5N1 in migratory birds and their potential role in virus spread, 205 pools of fecal samples from live migratory birds were analyzed. Here, the first virus isolation and genome characterization of two HPAIV H5N1 isolates from migratory birds (A/migratory bird/Bangladesh/P18/2010 and A/migratory bird/Bangladesh/P29/2010)are described. Full-length amplification, sequencing, and a comprehensive phylogenetic analysis were performed for HA, NA, M, NS, NP, PA, PB1, and PB2 gene segments. The selected migratory bird isolates belong to clade 2.3.2.1 along with recent Bangladeshi isolates from chickens, ducks, and crows which grouped in the same cluster with contemporary South and South-East Asian isolates. The studied isolates were genetically similar to other H5N1 isolates from different species within the respective clade although some unique amino acid substitutions were observed among them. Migratory birds remain a real threat for spreading pathogenic avian influenza viruses across the continent and introduction of new strains into Bangladesh.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Avian influenza viruses affect both domestic and wild bird populations all over the world. They often cause only mild infections in birds but can evolve into strains of high pathogenic nature causing high mortality in domestic bird populations [1]. The present outbreak of HPAI H5N1 virus began in 2003 and continued to spread in an unpredictable manner to almost all Asian countries as well as countries in Europe and Africa [2]. An isolate from China, Goose/Guangdong/1/96 [3], is considered as the progenitor H5N1 virus, from which the present panzootic H5N1 viruses have evolved [4–6]. The geographically large scale spread of HPAI H5N1 has been discussed to be associated with trade of poultry and poultry products as well as wild bird migration [7–10]. Isolation, characterization, and genome analysis of HPAI H5N1 virus from wild birds have been reported from several continents [11] and in countries like Hong Kong [12] and South Korea [13].The role of migratory birds in the spread of HPAI has also been highlighted.
Bangladesh attracts large numbers of wild birds overwinter in wetland habitat offered by the Ganges River and its tributaries. Every year more than 30 species have arrived in winter season in Bangladesh. Lesser Whistling Teal, Greater Whistling Teal, Cotton Pigmy Goose, Pochard, Darters (Snake bird), Pintail Duck, Herons, Comb Duck Gurgani, kingfishers, Egrets, Bitterns, Storks, and Flycatche are some of them. The country is situated on the cross roads of two major flyways for migratory birds, which is the southeastern end of the Central Asian Flyway, and the southwestern end of the East Asian—Australasian flyway. In addition, corridors for migratory birds extend to Russia at its northern limit [14]. Bangladesh first experienced HPAI outbreaks in poultry in March 2007 [15–17]. Since the first outbreak, the HPAI (H5N1) virus has spread to at least 54 districts and forced the authority to cull more than 2.5 million chickens of 779 affected units including 722 commercial and 57 backyard farms. So far, seven human cases of avian influenza with one death have been confirmed in Bangladesh [18]. Several studies have indicated that migratory birds might have played a significant role in the epidemiology and ecology of HPAI (H5N1) outbreaks in Bangladesh [19–21]. Factors likely to explain this claim are the temporal and spatial matching of HPAI (H5N1) outbreaks in domestic poultry and migratory bird’s activity in Bangladesh [20, 22]. However, evidence on the presence of virus in the migratory birds was felt necessary to clarify circumstantial evidences generated earlier [20].
In the present study, the first isolation and genome characterization of HPAI H5N1 virus from migratory birds in Bangladesh space are described. The provided information will help to better understand the molecular epidemiology of HPAI H5N1 in migratory and domestic birds in Bangladesh.
Methods
Sample collection and virus isolation
The preliminary sample collection and virus isolation were done at the National Reference Laboratory for Avian Influenza at Bangladesh. Subsequently, the molecular characterization was done at the Institute of Virology,University of Leipzig Germany. The samples were collected during the winter season (11/2009 to 03/2010 and 11/2010 to 03/2011) near the lakes at Bangladesh Livestock Research Institute (BLRI) and Jahangirnagar University (JU), Savar, Dhaka, situated in the central part of the country. Fecal samples were collected from fresh voids of migratory birds on the grass blade or in the bank of the lake where they rest overnight. Total 2,050 fecal samples were collected and transferred in ice pack, approximately 1 g fecal samples of each of 10 birds were pooled, mixed well, and placed in a 15 ml sterile falcon tube containing 3 ml isotonic solution consisting of phosphate buffer saline (PBS), pH 7.2, 50 % glycerol, penicillin (10,000 U/ml), Gentamycin (250 μg/ml), and Nystatin (2,500 U/ml). The samples were centrifuged at 3,000 rpm for 15 min and supernatants after collection inoculated into embryonated eggs (≈10 days) through the allantoic route. The eggs were incubated at 37 °C and checked daily for embryonic death until day 6 post inoculation; two serial passages in embryonated chicken eggs were performed before the sample was considered negative. The eggs allantoic fluid (AF) was collected irrespective of embryo mortality and subjected to hemagglutination test.
Genome amplification and sequencing
The hemagglutination positive samples were further tested by the RT-PCR for confirmation of the presence of avian influenza virus and its subtype. RNA was extracted using Qiagen RNeasy Extraction Kit, and RT-PCR was performed with the Qiagen One Step RT-PCR Kit (Hilden, Germany). Previously described primer pairs specific for the M gene were used for confirmation type A AIV [23]. The subtype specificity of AIV H5 and N1 was performed with the primers previously described [24, 25]. RT-PCR was done for the full-length amplification of 8 genome segments of confirmed H5N1 viruses using previously described protocol [26] with slight modification. Reverse transcription was done under standard condition of Revert Aid Reverse Transcriptase (Thermo Scientific, Germany) using the Uni 12 primer. PCR products were purified after agarose gel electrophoresis (GeneJET Gel Extraction Kit, Thermo Scientific, Germany) and the purified PCR products were used for direct nucleotide sequencing using Rhodamin Dye-Terminator Cycle Sequencing Ready Reaction Kit (Big Dye® Terminator v1.1; Applied Biosystem), followed by analysis in an ABIPRISM™ 310 genetic Analyser (Applied Biosystems).
Phylogeny and molecular genetic analyses
The full-length nucleotide sequences of the hemagglutinin (HA), neuraminidase (NA), matrix (M), non-structural (NS), nucleoprotein (NP) gene and partial nucleotide sequence of polymerase gene (PA [1985nt], PB1[1857nt], and PB2[1963nt]) of two isolates from migratory birds were subjected to Clustal W multiple sequence alignment as well as residue analyses using BioEdit 7.1.5 program. Assembled sequences were then blasted using the NCBI database for sequence confirmation and other available sequences. Additional influenza viruses representing each clade of HA gene [27, 28] available in GenBank were downloaded for comparative study. Nucleotide sequence identity and distance were calculated to check the homology between migratory birds isolate and compared with other selected reference isolates. Phylogenetic analyses were carried out by distance based neighbor-joining (NJ) method with 1,000 bootstrap replicates using software MEGA 5.10 version. Maximum Composite Likelihood substitution model was used for each phylogenetic analysis. The strains in each phylogenetic tree were chosen on basis of (i) their sequence homology (ii) geographic origin, (iii) year of isolation in order to follow the genetic evolution, and (iv) the WHO and FAO recommendations on representative H5 strains from the particular clades. The nucleotide (nt) sequences obtained from this study are available at GenBank under the accession numbers KJ397915 to KJ397924.
Results
Virus isolation and identification
The collected total 205 pools of fecal samples from live migratory birds were inoculated into embyronated chicken eggs. Laboratory analysis of AF post inoculation showed that 16 out of 205 pooled samples contained hemagglutinating agents. Among the hemagglutination positive samples 4 were AIV type A positive on M gene RT-PCR. Of which, two samples of migratory birds were positive for H5 and N1 subtype, named A/migratory bird/Bangladesh/P18/2010 (P18) and A/migratory bird/Bangladesh/P29/2010 (P29). Further full-length amplification of two surface glycoprotein genes (HA and NA) and six internal gene segments (M, NS, NP, PA, PB1, and PB2) were accomplished at their respective genome length.
Phylogenetic analyses
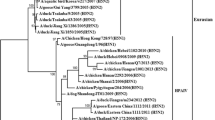
To understand the evolutionary history and origin of the selected isolates from live migratory birds, phylogenetic analysis of HA, NA, and each of 6 internal gene segments was conducted. Phylogenetic analysis based on the HA gene confirmed that the selected isolates belonged to clade 2.3.2.1 and they clustered with contemporary isolates from Bangladesh, India, Nepal, China, Japan, and Mongolia from different types of birds including domestic chickens (Fig. 1). The reported isolates in Bangladesh poultry from 2007 to 2010 were Qinghai-like, and belonged to clade 2.2.2. Whereas the P18 and P29 together with recent Bangladeshi isolate (2011 till date, reported in Genbank) from chickens, ducks, and crows belonged to clade 2.3.2.1. When clade 2.3.2.1 was observed intensively (Fig. 1), all selected Bangladeshi recent isolates including P18 and P29 along with some Indian H5N1 formed a broad cluster. The defined cluster rooted on A/Hubei/1/2010-like (HB) among the two other representatives of the respective clade reported by FAO. It was found that, these isolates form HB subgroup shared 99 % nt homology. The NA analyses revealed, P18 and P29 clustered together with isolates of clade 2.3.2.1 in the same group and made a clear distance from isolates of clade 2.2.2. The Bangladeshi isolates of clade 2.3.2.1 showed nearest relation with Indian isolates in the same group. A close relation was observed with isolates of ducks and environment from China and Laos forming another subgroup within the respective clade (Fig. 2).

Phylogenetic relationship and clade description of the HA gene of the selected isolates along with representative H5 avian influenza viruses. Numbers at the branch nodes indicate neighbor-joining bootstrap values ≥70 %. Analyses were based on 1,709 nucleotide and the tree rooted to A/goose/Guangdong/1/1996 which indicated green in color. Clade numbers refer to WHO H5N1 clade designations (27, 28). Selected isolates from migratory birds are marked with red round and the taxon name showed red in color. Other colors represent the group leader of their respective clades

Phylogenetic relationship of the NA gene of the selected isolates along with representative H5 avian influenza viruses. Numbers at the branch nodes indicate neighbor-joining bootstrap values ≥70 %. Analyses were based on 1,342 nucleotide and the tree rooted to A/goose/Guangdong/1/1996 which indicated green in color. Selected isolates from migratory birds are marked with red round and the taxon name showed red in color
The phylogenetic tree of six internal genes (M, NS, NP, PA, PB1, and PB2) of the two studied migratory birds revealed a very close evolutionary relationship with other isolates from Bangladesh and India forming a separate cluster (Fig. 3a–f). The two migratory birds isolate shared 99.2–100 % nucleotide homology in between themselves in respective to all internal genes. The tree-maintained progeny relation to a separate cluster with isolates of Lao, Vietnam, Mongolia, Cambodia, and China which are also member of clade 2.3.2.1. Internal genes of this defined clade isolates from chicken, duck, and environment share 98–100 % of nucleotide identity (Electronic Supplementary Material 1). Also they showed a clear distance from A/chicken/Afghanistan/1207/2006, a representative member of EMA2 (clade 2.2) and from the root A/Goose/Guangdong/1/1996.


a–f Phylogenetic relationship of M (a), NS (b), NP (c), PA (d), PB1 (e), and PB2 (f) gene of the migratory bird isolates along with representative influenza viruses. Numbers at the branch nodes indicate neighbor-joining bootstrap values ≥70 %. Analyses were based on nucleotide sequences (complete or partial) and all internal gene trees rooted to A/goose/Guangdong/1/1996 which indicated green in color. Selected isolates from migratory birds are marked in round and red in color
Amino acid comparison
Multiple alignment of deduced amino acid (aa) sequences of the viruses P18, P29 and selected Asian isolates were performed to characterize the amino acid differences within the proteins HA, NA, M, NS, NP, PA, PB1, and PB2.
In the HA alignment, P18 and P29 had five aa substitutions between themselves (Table 1) and eleven unique substitutions were recorded when compared with other selected Bangladeshi isolates of both clade reported in GenBank (Table 2). Both characterized viruses had multi-basic QRERRRK-R*G cleavage site in the HA molecule, which is the marker of high pathogenicity. Almost all 2.3.2.1 clade viruses included in this study had the same cleavage motif. Whereas the previous Bangladeshi isolates of clade 2.2.2 (Qinghai-like) from 2007 to 2008 along with others isolates of the same clade showed one additional basic amino acid (Table 3). However, the receptor binding pocket of the HA1 molecule of analyzed H5N1 viruses retained amino acid residues 222Q and 224G (H5 numbering) indicating preferential binding to α-2-3-Gal receptors. The two studied isolates and the other recent Bangladeshi isolates of clade 2.3.2.1 had an S129L (serine to leucine) substitution in their receptor binding site (Table 3). Although the effect of the substitution on virus replication was unknown. Details of HA aa substitutions are also available as Electronic Supplementary Material 2. The aa sequence analysis of NA, revealed no differences between P18 and P29 (Table 1). Both viruses showed a 20 aa stalk deletion in the NA protein when compared to the progenitor virus A/goose/Guangdong/1/1996 (Table 3).
The aa sequences for the five internal genes (M, NS NP, PA, PB1, and PB2) were similar between the viruses P18 and P29. They showed the highest (99–100 %) homologies to other’s H5N1 isolates from domestic poultry along with more than 300 H5N1 viruses from different countries of the continent Asia (Bangladesh, India, Thailand, Vietnam, Lao, Cambodia, and China) including a broad species difference. No amino acid mutation was observed in the conserved regions of internal protein related to host viral interaction, pathogenesis, or increase virulence. Thus, polymerase (PA, PB1, and PB2) gene data were not included in Table 3. So, the internal genes were found to be highly conserved among a wide range of isolates from the Continent in Asia.
Discussion
From 2007 through 2010, viruses of clade 2.2 caused outbreaks in Bangladesh and became endemic [29]. In 2011, viruses of two other clades (clade 2.3.2 and clade 2.3.4) were introduced in Bangladesh and detected in ducks, quails, chickens, and feral crows [30]. HPAI H5N1 viruses of Clade 2.3.2 were first identified in China and Vietnam at 2005 from ducks, geese, and other mammals [31, 32]. Since 2006–2007, H5N1 viruses of clades 2.3.2 and 2.3.4 were predominantly isolated from wild birds in Hong Kong [12, 33]. Although several studies indicated, based on circumstantial epidemiological evidences, wild migratory birds as key determinants for HPAI (H5N1) epidemiology, no viruses were isolated from wild migratory birds in Bangladesh [20, 21]. The present study demonstrated for the first time the presence of HPAI (H5N1) in the wild migratory bird population in Bangladesh.
FAO-OIE-WHO suggested a recent increase of HPAI (H5N1) outbreaks due to clade 2.3.2.1 in south and south-east Asia. So far, viruses of same clade were reported from India, Lao, Vietnam, Nepal, China, Hong Kong, Russia, Japan, and Bangladesh [30–35]. In some countries like Vietnam, clade 2.3.2.1 viruses are replacing the other clades completely [30]. Although clade 2.3.2.1 has been co-circulating with clade 2.2 and clade 2.3.4 [30], it is not unlikely that clade 2.3.2.1 could supercede other clades in Bangladesh. The clade 2.3.2.1 viruses were found to be significantly different in their antigenic properties [35].
The phylogenetic and molecular genetic analysis of the mentioned gene segments of the studied two isolates from migratory birds revealed genetic similarities with clade 2.3.2.1 viruses currently circulating among domestic poultry in Bangladesh. Three variants of clade 2.3.2.1 namely; A/Hubei/1/2010-like (HB), 2.3.2.1 A/barn swallow/Hong Kong/D10-1161/2010-like (BS), and 2.3.2.1 A/Hong Kong/6841/2010-like (HK), during 2010-2012 in Asia providing evidence for the continued incursion of new viruses or the evolution of existing viruses into Southeast Asia [36]. Although some unique amino acid substitutions in the HA gene were observed, none of those substitutions have direct relations with the conserved regions involved in molecular pathogenesis or host pathogen interactions. Detailed pathogenesis studies are necessary to understand the function of those substitutions. The genetic analysis from the migratory bird isolates strongly suggests that bird migration contributes to the spread of clade 2.3.2.1 in Asia. According to the epidemiology, history of origin and genetic analysis exchange by migratory birds mostly appear to be intra-continental. Evidence of intercontinental virus exchange is limited. It was reported from a surveillance study in Canada and United States [37]. The present study provides virological evidence for and also highlights the role of migratory birds in the expansion of geographical distribution of avian influenza virus not only in Bangladesh but also over South East Asia. However, this study independently is unable to verify whether wild birds are carriers or victims. Interestingly, the migratory bird samples that were found positive for clade 2.3.2.1 HPAI (H5N1) were obtained even earlier than the first detection of similar viruses in chicken, ducks, quails, crows, and environment in Bangladesh. The results of the present study, therefore, suggest that migratory birds are an important source of introduction of new HPAI (H5N1) virus strains into Bangladesh. Extensive virological surveillance is required to further explore the role of migratory birds in the epidemiology of HPAI.
Conclusion
Presence of the HPAI (H5N1) virus in wild migratory birds substantiates early circumstantial evidences, generated by different predictive epidemiological studies, that bird migration plays an important role in the introduction of HPAI (H5N1) viruses in Bangladesh. Thus, extensive surveillance in migratory birds along with described analysis of their flyways over Bangladesh would help to clarify their role in the introduction of new viruses in Bangladesh.
References
WHO, Avian Influenza A (H5N1) in Humans and Poultry in Viet Nam, Jan 13 (2004), http://www.who.int/csr/don/2004_01_13/en/
OIE, OIE Manual of diagnosis tests and vaccines for terrestrial animals, Chap. 2.3.4 (OIE, Paris, 2008)
X. Xu, K. Subbarao, N.J. Cox, Y. Guo, Genetic characterization of the pathogenic influenza A/Goose/Guangdong/1/96 (H5N1) virus: similarity of its hemagglutinin gene to those of H5N1 viruses from the 1997 outbreaks in Hong Kong. Virology 261, 15–19 (1999)
K.S. Li, Y. Guan, J. Wang, G.J.D. Smith, K.M. Xu, L. Duan, A.P. Rahardjo, P. Puthavathana et al., Genesis of a highly pathogenic and potentially pandemic H5N1 influenza virus in eastern Asia. Nature 430, 209–213 (2004)
FAO, H5N1 HPAI, Global overview, February 2010. Prepared by EMPRES/FAO-GLEWS, Issue no. 20. (2010), http://www.fao.org/docrep/012/ak737e/ak737e00.pdf
The World Health Organization Global Influenza Program Surveillance Network. Evolution of H5N1 Avian Influenza Viruses in Asia. Emerging Infectious Diseases http://www.cdc.gov/eid; Vol. 11, No. 10. (2005), http://wwwnc.cdc.gov/eid/article/11/10/pdfs/05-0644.pdf
Y. Guan, J.S. Peiris, A.S. Lipatov, T.M. Ellis, K.C. Dyrting, S. Krauss, L.J. Zhang, R.G. Webster, K.F. Shortridge, Emergence of multiple genotypes of H5N1 avian influenza viruses in Hong Kong SAR. Proc. Natl. Acad. Sci. U.S.A. 99, 8950–8955 (2002)
H. Chen, G.J.D. Smith, S.Y. Zhang, K. Qin, J. Wang, K.S. Li, R.G. Webster, J.S.M. Peiris, Y. Guan, H5N1 virus outbreak in migratory waterfowl. Nature 436, 191–192 (2005)
A.K. Chakrabarti, S.D. Pawar, S.S. Cherian, S.S. Koratkar, S.M. Jadhav, B. Pal, S. Raut, V. Thite, S.S. Kode, S.S. Keng, B.J. Payyapilly, J. Mullick, A.K. Mishra, Characterization of the influenza A H5N1 viruses of the 2008–2009 outbreaks in India reveals a third introduction and possible endemicity. PLoS ONE 4(11), e7846 (2009). doi:10.1371/journal.pone.0007846
C. Tosh, S. Nagarajan, H.V. Murugkar, R. Jain, P. Behera, M. Katare, D.D. Kulkarni, S.C. Dubey, Phylogenetic evidence of multiple introduction of H5N1 virus in Malda district of West Bengal India in 2008. Vet. Microbiol. 148, 132–139 (2011)
S.L. Salzberg, C. Kingsford, G. Cattoli, D.J. Spiro, D.A. Janies, M.M. Aly, I.H. Brown, E. Couacy-Hymann, G.M. De Mia, D.H. Dung et al., Genome analysis linking recent European and African influenza (H5N1) viruses. Emerg. Infect. Dis. 13, 713–718 (2007)
G.J. Smith, D. Vijaykrishna, T.M. Ellis, K.C. Dyrting, Y.H. Leung, J. Bahl, C.W. Wong, H. Kai, M.K. Chow, L. Duan, A.S. Chan, L.J. Zhang, H. Chen, G.S. Luk, J.S. Peiris, Y. Guan, Characterization of avian influenza viruses A (H5N1) from wild birds, Hong Kong, 2004–2008. Emerg. Infect. Dis. 15, 402–407 (2009)
H.I. Kwon, Song, P.N. Pascua, Y.H. Baek, J.H. Lee, S.P. Hong, J.B. Rho, J.K. Kim, H. Poo, C.J. Kim, Y.K. Choi, Genetic characterization and pathogenicity assessment of highly pathogenic H5N1 avian influenza viruses isolated from migratory wild birds in 2011, South Korea. Virus Res. 160((1–2)), 305–315 (2011). doi:10.1016/j.virusres.2011.07.003
B. Olsen, J.V. Munster, A. Wallensten, J. Waldenström, A. Osterhaus, R. Fouchier, Global patterns of influenza A virus in wild birds. Science 312(5772), 384–388 (2006)
WHO, H5N1 avian influenza: Timeline of major events. (2012), http://www.who.int/csr/disease/avian_influenza/H5N1_avian_influenza_update.pdf
P.K. Biswas, J.P. Christensen, S.S. Ahmed, H. Barua, A. Das, M.H. Rahman, M. Giasuddin, A.S. Hannan, M.A. Habib, A. Ahad, A.S. Rahman, R. Faruque, N.C. Debnath, Avian influenza outbreaks in chickens, Bangladesh. Emerg. Infect. Dis. 14, 1909–1912 (2008)
M.R. Islam, M. Giasuddin, M.A. Samad, M.J.F.A. Taimur, M.M. Alam, M.A. Baqi, A.T.M. Mahbub-E-Elahi, M.M. Amin, Phylogenetic analysis of highly pathogenic avian influenza (H5N1) virus isolates of first two waves in Bangladesh. Bangladesh J. Livest. Res. 15, 1–6 (2008)
WHO/GIP data in HQ as of 10 December. (2013), http://www.who.int/influenza/human_animal_interface/EN_GIP_20131210CumulativeNumberH5N1cases.pdf?ua=1
S.S. Ahmed, A.K. Ersboll, P.K. Biswas, J.P. Christensen, The space–time clustering of highly pathogenic avian influenza (HPAI) H5N1 outbreaks in Bangladesh. Epidemiol. Infect. 138, 843–852 (2010)
S.S. Ahmed, A.K. Ersboll, P.K. Biswas, J.P. Christensen, N. Toft, Spatio-temporal magnitude and direction of highly pathogenic avian influenza (H5N1) outbreaks in Bangladesh. PLoS ONE 6, e24324 (2011)
S.S. Ahmed, A.K. Ersboll, P.K. Biswas, J.P. Christensen, A.S. Hannan, N. Toft, Ecological determinants of highly pathogenic avian influenza (H5N1) outbreaks in Bangladesh. PLoS ONE 7, e33938 (2012)
M. Gilbert, S.H. Newman, J.Y. Takekawa, L. Loth, C. Biradar, D.J. Prosser, S. Balachandran, M.V. Subba Rao, T. Mundkur, B. Yan, Z. Xing, Y. Hou, N. Batbayar, T. Natsagdorj, L. Hogerwerf, J. Slingenbergh, X. Xiao, Flying over an infected landscape: distribution of highly pathogenic avian influenza H5N1 risk in South Asia and satellite tracking of wild waterfowl. EcoHealth (2011). doi:10.1007/s10393-10010-10672-10398
R.A. Fouchier, T.M. Bestebroer, S. Herfst, L. Van Der Kemp, G.F. Rimmelzwaan, A.D. Osterhaus, Detection of influenza A viruses from different species by PCR amplification of conserved sequences in the matrix gene. J. Clin. Microbiol. 38, 4096–4101 (2000)
M.S. Lee, P.C. Chang, J.H. Shien, M.C. Cheng, H.K. Shieh, Identification and subtyping of avian influenza viruses by reverse transcription-PCR. J. Virol. Methods 97, 13–22 (2001)
WHO, Recommendations and laboratory procedures for detection of avian influenza A (H5N1) virus in specimens from suspected human cases. (2007), http://www.who.int/csr/disease/avian_influenza/guidelines/RecAIlabtestsAug07.pdf
E. Hoffmann, J. Stech, Y. Guan, R.G. Webster, D.R. Perez, Universal primer set for the full-length amplification of all influenza A viruses. Arch. Virol. 146, 2275–2289 (2001)
WHO, OIE, FAO, Toward a unified nomenclature system for highly pathogenic avian influenza virus (H5N1). Emerg. Infect. Dis. 14, e1 (2008)
WHO/OIE/FAO H5N1 Evolution Working Group, Continued evolution of highly pathogenic avian influenza A (H5N1): updated nomenclature. Influenza and Other Respiratory Viruses, 6, 1–5 (2012). doi:10.1111/j.1750-2659.2011.00298.x, http://onlinelibrary.wiley.com/doi/10.1111/j.1750-2659.2011.00298.x/abstract
S.S. Ahmed, G.E. Themudo, J.P. Christensen, P.K. Biswas, M. Giasuddin, M.A. Samad, N. Toft, A.K. Ersboll, Molecular epidemiology of circulating highly pathogenic avian influenza (H5N1) virus in chickens, in Bangladesh, 2007–2010. Vaccine 30, 7381–7390 (2012)
M.R. Islam, M.E. Haque, M. Giasuddin, E.H. Chowdhury, M.A. Samad, R. Parvin, M. Nooruzzaman, M.M. Rahman, P. Monoura, New introduction of clade 2.3.2.1 avian influenza virus (H5N1) into Bangladesh. Transbound. Emerg. Dis. 59, 460–463 (2012)
H. Chen, G.J. Smith, K.S. Li, J. Wang, X.H. Fan, J.M. Rayner, D. Vijaykrishna, J.X. Zhang, L.J. Zhang, C.T. Guo, C.L. Cheung, K.M. Xu, L. Duan, K. Huang, K. Qin, Y.H. Leung, W.L. Wu, H.R. Lu, Y. Chen, N.S. Xia, T.S. Naipospos, K.Y. Yuen, S.S. Hassan, S. Bahri, T.D. Nguyen, R.G. Webster, J.S. Peiris, Y. Guan, Establishment of multiple sublineages of H5N1 influenza virus in Asia: implications for pandemic control. Proc. Natl. Acad. Sci. U.S.A. 103, 2845–2850 (2006)
S.I. Roberton, D.J. Bell, G.J. Smith, J.M. Nicholls, K.H. Chan, D.T. Nguyen, P.Q. Tran, U. Streicher, L.L. Poon, H. Chen, P. Horby, M. Guardo, Y. Guan, J.S. Peiris, Avian influenza H5N1 in viverrids: implications for wildlife health and conservation. Proc. Biol. Sci. 273, 1729–1732 (2006)
T.M. Ellis, K.C. Dyrting, C.W. Wong, B. Chadwick, C. Chan, M. Chiang, C. Li, P. Li, G.J. Smith, Y. Guan, J.S.M. Peiris, Analysis of H5N1 avian influenza infections from wild bird surveillance in Hong Kong from January 2006 to October 2007. Avian Pathol. 38, 107–119 (2009)
H.R. Kim, B.S. Kim, Y.C. Bae, O.K. Moon, J.K. Oem, H.M. Kang, J.G. Choi, O.S. Lee, Y.J. Lee, H5N1 subtype highly pathogenic avian influenza virus isolated from healthy mallard captured in South Korea. Vet. Microbiol. 151, 386–389 (2011)
S. Nagarajan, C. Tosh, D.K. Smith, J.S. Peiris, H.V. Murugkar, R. Sridevi, M. Kumar, M. Katare, R. Jain, Z. Syed, P. Behera, C.L. Cheung, R. Khandia, S. Tripathi, Y. Guan, S.C. Dubey, Avian influenza (H5N1) virus of clade 2.3.2 in domestic poultry in India. PLoS ONE 7, e31844 (2012)
FAO, Update on the continuous spread and expansion of H5N1 highly pathogenic avian influenza, Clade 2.3.2.1 in Asia (2010–2012). Focus on 7 (2014), http://www.fao.org/docrep/019/i3610e/i3610e.pdf
S. Krauss, C.A. Obert, J. Franks, D. Walker, K. Jones et al., Influenza in migratory birds and evidence of limited intercontinental virus exchange. PLoS Pathog. 3(11), e167 (2007). doi:10.1371/journal.ppat.0030167
Acknowledgments
This work was supported by German Academic Exchange Service (DAAD). We would like to thank Bangladesh Livestock Research Institute (BLRI), Savar, Dhaka, Bangladesh for initial funding as the part of research done at Bangladesh.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
11262_2014_1118_MOESM1_ESM.docx
Supplementary material 1 (DOCX 18 kb). Percent nucleotide homology comparison of the migratory birds isolates with other representatives H5N1
11262_2014_1118_MOESM2_ESM.pdf
Supplementary material 2 (PDF 51 kb). Amino acid substitutions in HA gene between two migratory birds isolate and with other representatives H5N1
11262_2014_1118_MOESM3_ESM.xlsx
Supplementary material 3 (XLSX 17 kb). A list of virus strains used in this study with GenBank accession number in respective to their all gene segments
Rights and permissions
About this article

Cite this article
Parvin, R., Kamal, A.H.M., Haque, M.E. et al. Genetic characterization of highly pathogenic H5N1 avian influenza virus from live migratory birds in Bangladesh. Virus Genes 49, 438–448 (2014). https://doi.org/10.1007/s11262-014-1118-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-014-1118-0