
Abstract
The complete nucleotide and deduced amino acid sequences of the RNA genome of a recently isolated norovirus (NoV) from Korea, designated Hu/GII-4/CBNU2/2007/KR (CBNU2), were determined and characterized by phylogenetic comparison with several genetically diverse NoV sequences. The RNA genome of CBNU2 is 7,560 nucleotides in length, excluding the 3′ poly (A) tract. It includes three open reading frames (ORFs): ORF1, which encodes the nonstructural polyprotein (5–5,104); ORF2, which encodes VP1 (5,085–6,707); and ORF3, which encodes VP2 (6,707–7,513). ORF2-based phylogenetic analysis revealed that CBNU2 belonged to the GII.4 genotype, the most prevalent genotype, and formed a cluster with NoVs isolated from Asian regions, between 2006 and 2008. Comparative analysis with the consensus sequence of 207 completely sequenced NoV genomes showed 47 mismatched nucleotides: 26 in ORF1, 14 in ORF2, and 7 in ORF3, resulting in 8 amino acid changes: 3 in ORF1, 2 in ORF2, and 3 in ORF3. Phylogenetic analysis with full genome ORF1, ORF2, and ORF3 nucleotide sequences obtained from CBNU2 and each of the other representative NoV genomes suggested that CBNU2 had not undergone recombination with any of the other NoVs. A SimPlot analysis further supported this finding.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Noroviruses (NoVs) are members of the Caliciviridae, with positive-sense single-stranded RNA genomes ~7.5 kb in length [1]. The full genome of NoV consists of three open reading frames (ORFs), with a poly-A tail at the 3′-end and with a single 10–20 bp repeat at the 5′-end, near the 5,100th base [2].
More than 90 % of non-bacterial gastroenteritis is caused by NoV. Infection with NoV generally includes a latent period of 24–48 h, followed by active infection with typical gastroenteritis symptoms such as nausea, stomachache, diarrhea, and fever [3, 4]. NoVs are spread through person-to-person contact, via the fecal–oral route, and through polluted water and food. As NoV strains are diverse, co-infection with two or more types of NoV is often observed. Also, only a small amount of NoV is required for infection, resulting in continuous reports of outbreaks at schools, hospitals, and nursing homes [5–7]. In Korea, a large outbreak caused by such NoV has recently been declared, and in response, the characteristics of NoVs in Korea have been investigated. In addition, a number of studies on planning, monitoring, and response systems have been conducted [8–18].
Noroviruses have been classified into five genogroups (GI–GV). Of these, GI, GII, and GIV have been shown to be infectious to humans. Epidemiological investigations have shown that the GII.4 group is detected in more than 90 % of disease cases, is actively propagated, and has been broadly detected in Korea [16, 19, 20]. A general method for genotyping NoVs was proposed by Zheng et al. [21]. In this method, the full length ORF2 sequence, including the ORF1/2 junction, is used. This represents a limitation in determining the genotype of the virus, however, since only a partial nucleotide sequence is analyzed rather than the full sequence, resulting in increased reporting of genetic recombinants of NoVs [8, 22–25]. Thus, verification of the characteristics of the virus based on analysis of the entire nucleotide sequence would be helpful for further accurate analyses. Through the accumulation of a comprehensive set of whole-genome sequence data, a more accurate method for determining genotypes can be developed. In Korea, the first analysis of the full genome sequence of the CBNU1 strain, belonging to the GII.3 genotype, was performed in 2010 [18], and the CUK-3 strain was first reported as the GII.4 genotype in 2011 [16].
Here, we present the complete nucleotide sequence of the third human NoV genome in Korea to be sequenced in its entirety and the second GII.4-genotype NoV from Korea. This sequence was then used as the basis for various phylogenetic analyses, together with the nucleotide sequences of other members of the GII.4 genotype. The GII.4 NoVs are of particular concern due to the predominance of this group in human disease cases in recent times.
Materials and methods
Samples, RT-PCR, and sequencing
A stool sample from a 1-year-old patient suffering from acute gastroenteritis was kindly provided from Sanngye-Paik Hospital in 2007. The patient was diagnosed with NoV infection, with no other bacterial or viral infections known to cause acute gastroenteritis. The NoV was from the GII genogroup. After homogenizing in phosphate-buffered saline, the sample was centrifuged at 13,000 rpm, for 20 min, at 4 °C. The supernatant was diluted 1:50 using serum-free Dulbecco’s Modified Eagle Medium (DMEM; Cambrex Bio Science Walkersville, Inc., Walkersville, MD). An easy-red total RNA extract kit (iNtRON Biotechnology, Sunggnam, Korea) was used to extract 30 μl of RNA from 200 μl of supernatant, following the manufacturer’s protocol. The reverse-transcription (RT) reaction was performed using a Sensiscript RT kit (Qiagen, Hilden, Germany), following the manufacturer’s protocol. In brief, 3 μl of RNA template, 2 μl of Buffer RT (10×), 2 μl of dNTP Mix (5 mM each), 2 μl of reverse primer (10 pmol), and 1 μl of Sensiscript Reverse Transcriptase were added to 10 μl RNase-free water to give a final volume of 20 μl. The RT reaction was carried out at 37 °C for 60 min and 94 °C for 3 min. Four reverse primers were applied for RT: GII-4-r908, GII-4-MR1, COG2R, and Tx30SXN (Table 1). The total reaction volume was 50 μl, composed of 2 μl cDNAs, 25 μl 2× PCR Master mix solution (i-star MAXII, iNtRON Biotechnology) including a mixture of Taq polymerase and a DNA polymerase with proofreading activity (error rate, 1.8 × 10−8), 1 μl (10 pmol) of each of the primers (forward and reverse, Table 1), and 21 μl nuclease-free water. Then, PCR was performed at 94 °C for 3 min followed by 30 cycles of 94 °C for 10 s, 60 °C for 30 s, and 72 °C for variable cycle times (the time was determined according to the length of each PCR product), and a final step at 72 °C for 8 min. A T Gradient PCR machine (Biometra, Göttingen, Germany) was used to carry out the RT-PCR. Five PCR products were analyzed by electrophoresis on a 1.5 % agarose gel, purified with Gel SV (GeneAll Biotechnology, Seoul, Korea) and cloned into the pGEM-T easy vector (Promega, Madison, WI). The nucleotide sequencing was conducted by Solgent (Daejeon, Korea) using a BigDye terminator v3.1 cycle sequencing kit (Applied Biosystems, Carlsbad, CA) and an ABI 3730XL DNA analyzer (Applied Biosystems).
Characterization of the full genome
Using a BLAST search of the National Center for Biotechnology Information (NCBI) database, three nucleotide sequences [Aichi3 (AB447446), Toyama4 (AB447444), and Fukui2 (AB541245)], which were similar to the full nucleotide sequences obtained in this study, were referenced. The positions of ORFs 1, 2, and 3 were verified using the CLC sequence viewer (version 6.0, http://www.clcbio.com/) and the Local BLAST and ORF finder of NCBI. In addition, the location of the ORFs was determined using BioEdit (version 7.1.3.0, http://www.mbio.ncsu.edu/bioedit/bioedit.html).
Phylogenetic analysis of the NoV CBNU2
The nucleotide sequences of the ORF2 region have served as the major basis for phylogenetic analysis of NoVs [21]. A total of 70 ORF2 sequences were used for the analysis: 68 NoV nucleotide sequences, identified by Zheng et al. in 2006 [21] and obtained via an NCBI search, the ORF2 sequence of CBNU1 reported by Yun et al. [18], and the sequences of the NoVs determined in this study (Table 2). Alignment of the sequences was done using ClustalX (version 2.0), and evolutionary distance was calculated using the PHYLIP package (version 3.69, http://cmgm.stanford.edu/phylip/) to yield a phylogenetic tree. In brief, the evolutionary distance between arranged sequences was calculated using the DNADIST program based on the Kimura two-parameter method, and the phylogenetic tree was determined using the NEIGHBOR program based on the neighbor-joining method. Then, the phylogenetic tree was expressed using the TreeView program (version 1.6.6). The significance of the phylogenetic trees was supported by bootstrap analysis. Phylogenetic trees were constructed from 1,000 replicates generated by the Seqboot program. The consensus tree was generated by the Consense program. In addition, we compared 207 nucleotide sequences from GII.4-genotype NoVs in order to further characterize CBNU2, in terms of molecular evolution and phylogeny within the GII.4 group. For this phylogenetic analysis, ORF2 nucleotide sequences were extracted from these 207 complete genome sequences (GenBank accession numbers AB447427–447463, AB541201–AB541220, AB541222–AB541299, AB541301–AB541361, AB543808, DQ078814, DQ658413, FJ514242, FJ537134–537138, GQ845367, and HQ009513), and phylogenetic analyses were carried out in the same manner as described above. The 207 sequences were selected from the GenBank database with selection criteria as follows: (1) they must be from GII.4-genotype NoVs and (2) they must be complete genome sequences.
Sequence alignment and comparative analysis of NoV genomes
The consensus sequence for the NoV GII-4 group was obtained using ClustalX and CLC sequence viewer, from a total of 207 NoV GII-4 whole-genome nucleotide sequences obtained as described above. Then, the consensus nucleotide sequence was compared with the full genome sequence of NoV CBNU2 obtained in this study. In addition, to investigate the difference between the full genome sequence of CBNU2 and the full genome sequence of the Korean isolate, CUK-3, the full genome sequences of CUK-3 and NoV CBNU2 were compared. For this comparison, the sequences were aligned using ClustalX, and the mismatched sequences were verified using BioEdit. To determine the difference between the two amino acid sequences, the previously used nucleotide sequences were translated into amino acid sequences using the CLC sequence viewer and aligned using the ClustalX program. Mismatching was also verified using BioEdit.
Verification of recombination in CBNU2
Using the SeqMan program (DNASTAR, Madison, WI, USA), phylogenetic analyses were conducted after aligning the genome of CBNU2 and a representative NoV genome for each genogroup with the entire nucleotide sequence and subsequences for ORFs 1, 2, and 3. The reference sequences used were as follows: Norwalk (NC001959) was the reference sequence for GI; Newbury2 (AF097917) was the reference sequence for GIII; MNV-1 (AY228235) was the reference sequence for GV; Texas/TCH04-577 (AB365435), Saitama U18 (AB039781), and Saitama U201 (AB039782) were the reference sequences for GII.3 genotype; and Guanzhow/NVgz01 (DQ369797), Sakai2 (AB447448), and Chiba/04-1050 (AB220921) were the reference sequences for GII.4 genotype. The phylogenetic analyses were carried out in the same manner as described above. In addition, the similarities between these reference sequences, CBNU2, GII.4 (Aichi3, CUK3, Chiba/04-1050, Sakai2, and Guangzhou/NVgz01), GII.4 recombinant (CBNU1), and GII.3 (Texas/TCH04-577), were analyzed using SimPlot version 3.5.1, based on the Kimura-2 parameter distance model.
Results
Full genome sequencing of the CBNU2 NoV
Five PCR products from the CBNU2 genome were obtained using RT-PCR (Fig. 1a). The first RT-PCR product, obtained using the GII-4-F1 and GII-4-r908 primers, spanned nucleotides 1–929. The second RT-PCR product, obtained using the GII-4-MF1 and GII-4-MR1 primers, amplified a product spanning nucleotides 861–4,137. The third RT-PCR product, obtained using the N8 and COG2R primers, amplified a product spanning nucleotides 4,000–5,100. The fourth RT-PCR product, obtained using the JJV2F and COG2R primers, amplified a product spanning nucleotides 5,003–5,100. Finally, the nested PCR product, obtained using the JJVF2, G2SKF, and Tx30SXN primers, amplified a product spanning nucleotides 5,003–7,583 (Fig. 1b). The entire nucleotide sequence, 7,583 nucleotides in length, was constructed using a method for connecting overlapped sections in separate sequence runs as a single sequence, designated Norovirus Hu/GII-4/CBNU2/2007/KR (CBNU2) and submitted to GenBank (GenBank accession number, JQ622197). The ORF information was verified by ORF finding programs such as CLC Sequence Viewer (version 6.1 http://www.clcbio.com/index.php) and ORF finder provided by NCBI. In addition, three other nucleotide sequences [Aichi3 (AB447446), Toyama4 (AB447444), and Fukui2 (AB541245)], which were similar to the full nucleotide sequence obtained in this study, were referenced. Blast results using the CBNU2 nucleotide sequence as the query showed that query coverage was 98 % and maximum identity was 99 % with three reference strains in each operation (data not shown). The sequences spanning nucleotides 5–5,104, 5,085–6,707, and 6,707–7,513 represented ORF1, ORF2, and ORF3, respectively. Also, ORF1 and ORF2 overlapped over 20 nucleotides (nt 5,085–5,104) and ORF2 and ORF3 overlapped over a single nucleotide (nt 6,707) (Fig. 1). The poly-A tail of the CBNU2 NoV began at nt 7,561. Finally, the first 18 nucleotide sequences were repeated at position 5,081 (Fig. 1a).

Genome architecture of the NoV CBNU2. Nucleotides 5–5,104, 5,104–6,707, and 6,707–7,513 encode ORFs 1, 2, and 3, respectively. ORFs 1 and 2 share a 20-nt overlap, and ORFs 2 and 3 share a 1-nt overlap. A polyadenylation sequence extends from nt 7,561. The 18-nt sequence at the 5′-end of the genome is repeated at position 5,081
Phylogenetic analysis of NoV CBNU2
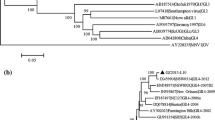
Phylogenetic analysis of CBNU2 was performed using the ORF2 sequence from 69 reference NoV sequences and the ORF2 of CBNU2 (see “Materials and methods” section). This analysis indicated that the GII genogroup could be classified into 17 genotypes. Of these, CBNU2 was verified as GII.4 genotype, which also included Bristol-GBR93, Fmhill-USA, and DG4770-USA. This genotype corresponds to the GII.4 genotype proposed by Zheng et al. [21] (Fig. 2a). Furthermore, genotyping was confirmed using the Norovirus Genotyping Tool version 1.0 developed by Kroneman et al. [26], which verified the GII.4 genotype for CBNU2 (data not shown). In addition, a phylogenetic analysis was performed using the ORF2 sequence of 207 other NoV GII.4 genotype members. This more focused analysis revealed four clusters within the GII.4 genotype, designated in this study as clusters A, B, C, and D. CBNU2 fell within cluster A, which has the largest number of NoV strains. Of these, 181 were isolated from Asian regions between 2006 and 2008 (Fig. 2b). Cluster B included 18 strains of NoV, isolated from Korea, North America, Australia, and Japan between 2004 and 2009. The five strains in cluster C were isolated from the USA before 1988. Finally, the four strains in cluster D were isolated from Osaka, Japan, in 2007 (Fig. 2b).

Phylogenetic analysis of CBNU2 and other NoVs. a Phylogenetic analysis using the ORF2 nucleotide sequences for representative strains for each genotype of NoV. Unrooted trees were shown to visualize phylogenetic relationships between the strains, which could be grouped into five main clusters: GI through GV. CBNU2 fell clearly into the GII.4 genotype, which included Bristol-GBR93, Fmhill-USA, and DG4770-USA. b Analysis using 207 GII.4 NoV ORF2 nucleotide sequences. The GII.4 genotype was divided into four clusters, A–D. CBNU2 belonged to cluster A, which included 181 strains isolated from Asia between 2006 and 2008 (circled and labeled in red). The significance of the phylogenetic trees was supported by bootstrap analysis of 1,000 replicates. Bootstrap values are shown on the corresponding branches (Color figure online)
Comparative analysis of CBNU2 and consensus sequences from completely sequenced NoV genomes
The ORF-encoding sequence of CBNU2, spanning nucleotides 5 through 7,513, was compared with the consensus sequences from the 207 NoV GII.4 genotypes. A total of 47 nucleotide sequences showed disagreements; 26, 14, and 7 in ORFs 1, 2, and 3, respectively (Table 3). Of these 47 nucleotide sequence differences, 39 caused no changes in the amino acid sequence. The remaining eight differences did result in amino acid sequence changes. Three of these were in ORF1, two were in ORF2, and the remaining three were in ORF3. The differences in nucleotide and amino acid sequences between CBNU2 and CUK-3, another NoV isolated in Korea, were also analyzed (Table 4). Because CUK-3 had a total of 7,559 reported nucleotides, CBNU2 nucleotides 1–7,559 were used for the comparison between these two strains. A total of 90 nucleotide differences were seen in the nucleotide sequences, with 57, 23, and 8 of these located in ORFs 1, 2, and 3, respectively, and two sequences in the 3′ untranslated region. No nucleotide sequence differences were seen in any areas of overlap between two ORFs. Although there were 90 disagreements at the nucleotide sequence level, only 17 of these resulted in changes in the amino acid sequence. These included nine, four, and four sequences in ORFs 1, 2, and 3, respectively, and are highlighted in Table 4.
Absence of recombination
The genome of CBNU2 was screened for recombination events by comparing individual sequence fragments with known sequence fragments from NoVs of different genotypes. Phylogenetic analyses were performed using representative NoV sequences for each genotype, obtained from NCBI. The sequences were divided into separate sequence fragments: the full genome, ORF1, ORF2, and ORF3, and aligned with the CBNU2 sequence. CBNU2 was in this way verified as non-recombinant, forming a group with the representative sequences of GII.4, such as Guanzhow/NVgz01 (DQ369797), Sakai2 (AB447448), and Chiba/04-1050 (AB220921), on the basis not only of the whole-genome sequence but also on the basis of analysis using single ORF sequences only (Fig. 3). In addition, on the basis of an analysis of similarity using SimPlot, CBNU2 and CUK-3 showed a very high similarity to Aichi3 (as a query), which is a GII.4 NoV, and the ORF sequences were very different from those of Texas/TCH04-577, which is a GII.3 NoV (Fig. 4). The SimPlot analysis also indicated that there was no genetic recombination in the CBNU2 genome. On the other hand, CBNU1, a recombinant between a GII.4 and a GII.3 NoV, was highly similar to GII.4 NoVs, such as Chiba/04-1050, Sakai2, and Guangzhou/NVgz01, in ORFs 1 and 2, but it was very similar to the GII.3 NoV Texas/TCH04-577 in ORF3 (Fig. 4).

Phylogenetic analyses based on the full genome sequences and three ORFs of representative strains for each genotype of NoV: a full genome sequences; b ORF1 nucleotide sequences; c ORF2 nucleotide sequences; and d ORF3 nucleotide sequences. The significance of the phylogenetic trees was supported by bootstrap analysis of 1,000 replicates. Bootstrap values are shown on the corresponding branches

Evidence for a lack of recombination in the CBNU2 genome. SimPlot analysis of the full genome of CBNU2 and those of seven reference strains [GII.4, Aichi3 (as a query), CUK3, Chiba/04-1050, Sakai2, and Guangzhou/NVgz01; GII.4 recombinant, CBNU1; GII.3, Texas/TCH04-577]. The SimPlot analysis employed the Kimura-2 parameter distance model with a window size of 200 bp and a step size of 20 bp with the gap strip on and was visualized as a similarity plot
Discussion
In this study, the full nucleotide sequence of the CBNU2 genome, a newly isolated strain from Korea, was characterized. It consisted of 7,583 nucleotides encoding three ORFs. All of them were oriented in the same polarity (positive stranded), with a 20-nt overlap between ORF1 and ORF2 and a 1-nt overlap between ORF2 and ORF3. Two other structural features that are similar to those of known NoVs are a poly-A sequence extending 3′ from nt 7,561 and an 18-bp sequence that is repeated at either end of ORF1 [2]. In addition, we determined via phylogenetic analysis that CBNU2 belongs to the NoV GII.4 genotype, which is dominant throughout the world [4, 5, 7, 19, 27–33]. In Korea, it has been known that the outbreak of acute gastroenteritis largely occurs due to NoVs during the winter season [11, 17], which in Korea generally extends from November to March. CBNU2 was first discovered in February of 2007 in Seoul, and CUK-3 was first isolated in November of 2008 in Daejeon [16]. The epidemiological patterns of infection of these viruses tend to be seasonal rather than regional. Though both the CBNU2 and CUK-3 strains were isolated in Korea and belong to the GII.4 genotype, there were clear differences in nucleotide and amino acid sequences between them. At the nucleotide and amino acid sequence levels, there were 90 and 17 differences, respectively. Comparison with consensus sequences of 207 reference strains in the GII.4 genotype revealed 47 nucleotide differences between CBNU2 and the GII-4 consensus sequence. These gave rise to eight amino acid differences, and this is in agreement with the general case of a higher level of conservation in ORFs 1 and 2 compared with ORF3 [31]. Among the 207 reference strains, two amino acid variations in ORF2 of CBNU2, N17S, and R411G, were also seen in AB541276 and AB541236. These two variations might induce conformational changes in the capsid protein of NoVs, because the ORF2 encodes the major capsid protein, VP1. Further studies are needed to elucidate such changes in ORF2, since this information would be helpful for developing a NoV vaccine. ORF3, which is generally poorly conserved, had three nucleotide differences for a total sequence length of only 807 nt, compared with the same number of nucleotide differences for 5,100 nt in ORF1 (six times the length of ORF3) and two nucleotide differences for 1,628 nt in ORF2.
A BLAST search using the CBNU2 sequence determined that Aichi3 showed the highest similarity, 99.41 %, to CBNU2, and verified the existence of several similar viruses to CBNU2, including 22 viruses that showed rates of similarity greater than 99 % (data not shown). In an analysis based on geographical region, of 100 highly similar viruses, 97 had been isolated from Japan, 1 from Korea and 2 from China. The large number of viruses isolated from Japan was not, however, considered to indicate a larger distribution and circulation of similar viruses to CBNU2; it probably instead simply reflects the fact that more viruses have been isolated and studied in Japan. Although no conclusions could be drawn regarding the prevalence of these viruses in Japan, China, and Korea, it is possible to infer that there are similar NoVs in these three East Asian countries. Similarity and close phylogenetic relationships between waterborne enteric viruses like NoVs in these countries have been previously reported [34–36]. Thus, it can be considered that the development of vaccines for the broadly distributed GII.4 NoVs could be applied to all three of these countries. Also, this shared geographical range emphasizes the importance of actively communicating information regarding future studies and joint research between these three countries.
A phylogenetic analysis focusing on the GII.4 NoVs, based on their ORF2 nucleotide sequences, revealed that the GII.4 genotype can be divided into four different clusters, A–D. The cluster group A, which included CBNU2, was constituted of 181 strains isolated from Asia between 2006 and 2008. The cluster group B included 18 strains isolated from Korea, America, Australia, and Japan between 2004 and 2009. The smallest clusters, groups C and D, included five strains isolated from America before 1988 and Osaka, Japan in 2007, respectively. This verified the existence of regional and chronological relationships in these four groups. It also revealed the state of continuous change over time among these viruses, while at the same time showing that the characteristics of viruses isolated during the same time period could vary according to geographical region.
The other strain isolated from Korea, CBNU1, was originally characterized as belonging to the GII.3 group, since it formed a group with Texas/TCH04-577, Saitama U18, and Saitama U201 in a phylogenetic analysis based on ORF2. Subsequent analysis, however, revealed that it is in fact a recombinant between a GII.4 and GII.3 viruses [18]. While the CBNU1 ORF2 clearly falls within the GII.3 genotype, an analysis based on ORF1 revealed that this sequence falls within the GII.4 genotype, with similarity to the sequences of Guanzhow/NVgz01, Sakai2, and Chiba/04-1050. The SimPlot analysis done for this study corroborates this characterization of CBNU1 (Fig. 4). In contrast, CBNU2 was clearly not a recombinant, since it fell within the GII.4 genotype, Guanzhow/NVgz01, Sakai2, and Chiba/04-1050, based not only on the ORF2 sequence, but also on the ORF1 sequence, the entire sequence, and even in a separate analysis based on the less-conserved ORF3. Continuing the analysis of possible recombination in Korean strains, the nucleotide sequences of CUK-3, which were isolated in Korea and belonged to the GII.4 genotype along with CBNU2, were also investigated. The results of this analysis indicated that CUK-3 is not a recombinant, forming a group with the representative viruses in the GII.4 genotype along with CBNU2 (Fig. 3). In addition, the results of an analysis of similarities using SimPlot verified that CBNU2 exhibited no evidence of genetic recombination among different strains; it showed a high similarity to the other GII.4 strains across its entire sequence (Fig. 4). Interestingly, CBNU2 showed a high similarity to CUK-3 and Aichi3 in the same SimPlot analysis, with a relatively low similarity to Guanzhow/NVgz01, Sakai2, and Chiba/04-1050 (Fig. 4). This reinforces the hypothesis that there exist clusters within the GII.4 genotype, with CBNU2, CUK-3, and Aichi3 belonging to cluster A and Sakai2 belonging to cluster B (Fig. 2b).
Recent years have seen an increase in reports of recombinants [8, 22–25], and limitations in classifying viruses into genogroups based on partial nucleotide sequences have presented a growing concern [5, 37, 38]. Thus, it is necessary to determine such genogroups with greater care, including analysis of entire nucleotide sequences in NoVs for more accurate phylogenetic classification. To date, most whole-genome NoV analyses have been performed in Japan. The present study confirms NoV CBNU2 as the third NoV isolated in Korea and the second Korean virus from the GII.4 genotype. Analysis of Korean strains represents an important issue in characterizing the NoVs of East Asia. It is expected that the results from this study will be useful for future genetic studies of NoVs both in Korea and throughout East Asia.
References
K.Y. Green, T. Ando, M.S. Balayan, T. Berke, I.N. Clarke, M.K. Estes, D.O. Matson, S. Nakata, J.D. Neill, M.J. Studdert, H.J. Thiel, J. Infect. Dis. 181(Suppl 2), S322–S330 (2000)
P.R. Lambden, E.O. Caul, C.R. Ashley, I.N. Clarke, Science 259, 516–519 (1993)
S. Inouye, K. Yamashita, S. Yamadera, M. Yoshikawa, N. Kato, N. Okabe, J. Infect. Dis. 181(Suppl 2), S270–S274 (2000)
M.C. Medici, M. Martinelli, L.A. Abelli, F.M. Ruggeri, I. Di Bartolo, M.C. Arcangeletti, F. Pinardi, F. De Conto, G. Izzi, S. Bernasconi, C. Chezzi, G. Dettori, J. Med. Virol. 78, 1486–1492 (2006)
R.L. Fankhauser, S.S. Monroe, J.S. Noel, C.D. Humphrey, J.S. Bresee, U.D. Parashar, T. Ando, R.I. Glass, J. Infect. Dis. 186, 1–7 (2002)
K.Y. Green, G. Belliot, J.L. Taylor, J. Valdesuso, J.F. Lew, A.Z. Kapikian, F.Y. Lin, J. Infect. Dis. 185, 133–146 (2002)
M.A. Widdowson, E.H. Cramer, L. Hadley, J.S. Bresee, R.S. Beard, S.N. Bulens, M. Charles, W. Chege, E. Isakbaeva, J.G. Wright, E. Mintz, D. Forney, J. Massey, R.I. Glass, S.S. Monroe, J. Infect. Dis. 190, 27–36 (2004)
J.Y. Chung, T.H. Han, S.H. Park, S.W. Kim, E.S. Hwang, J. Med. Virol. 82, 146–152 (2010)
T.H. Han, C.H. Kim, J.Y. Chung, S.H. Park, E.S. Hwang, Arch. Virol. 156, 323–329 (2011)
J.W. Huh, W.H. Kim, S.G. Moon, J.B. Lee, Y.H. Lim, J. Clin. Virol. 44, 152–156 (2009)
S.H. Kim, D.S. Cheon, J.H. Kim, D.H. Lee, W.H. Jheong, Y.J. Heo, H.M. Chung, Y. Jee, J.S. Lee, J. Clin. Microbiol. 43, 4836–4839 (2005)
V.P. Le, Y.C. Jung, K.S. Kang, I. Lim, S.C. Myung, W. Kim, J. Med. Virol. 82, 1065–1070 (2010)
S.G. Lee, S.H. Lee, S.W. Park, C.I. Suh, W.H. Jheong, S. Oh, S.Y. Paik, Virol. J. 8, 260 (2011)
S.G. Lee, W.H. Jheong, C.I. Suh, S.H. Kim, J.B. Lee, Y.S. Jeong, G. Ko, K.L. Jang, G.C. Lee, S.Y. Paik, Appl. Environ. Microbiol. 77, 1466–1474 (2011)
K.S. Park, H.S. Jeong, K.A. Baek, C.G. Lee, S.M. Park, J.S. Park, Y.J. Choi, H.J. Choi, D.S. Cheon, Arch. Virol. 155, 635–641 (2010)
J.W. Park, S.G. Lee, Y.M. Lee, W.H. Jheong, S. Ryu, S.Y. Paik, Virol. J. 8, 167 (2011)
J.S. Yoon, S.G. Lee, S.K. Hong, S.A. Lee, W.H. Jheong, S.S. Oh, M.H. Oh, G.P. Ko, C.H. Lee, S.Y. Paik, J. Clin. Microbiol. 46, 1474–1477 (2008)
S.I. Yun, J.K. Kim, B.H. Song, A.Y. Jeong, Y.M. Jee, C.H. Lee, S.Y. Paik, Y. Koo, I. Jeon, S.J. Byun, Y.M. Lee, Virus Res. 152, 137–152 (2010)
D.J. Allen, R. Noad, D. Samuel, J.J. Gray, P. Roy, M. Iturriza-Gómara, Virol. J. 6, 150 (2009)
T. Kageyama, S. Kojima, M. Shinohara, K. Uchida, S. Fukushi, F.B. Hoshino, N. Takeda, K. Katayama, J. Clin. Microbiol. 41, 1548–1557 (2003)
D.P. Zheng, T. Ando, R.L. Fankhauser, R.S. Beard, R.I. Glass, S.S. Monroe, Virology 346, 312–323 (2006)
K. Ambert-Balay, F. Bon, F. Le Guyader, P. Pothier, E. Kohli, J. Clin. Microbiol. 43, 5179–5186 (2005)
P. Chhabra, A.M. Walimbe, S.D. Chitambar, Virus Res. 147, 242–246 (2010)
S.K. Dey, T.G. Phan, M. Mizuguchia, S. Okitsua, H. Ushijima, Virus Genes 40, 362–364 (2010)
M. Jin, H.P. Xie, Z.J. Duan, N. Liu, Q. Zhang, B.S. Wu, H.Y. Li, W.X. Cheng, S.H. Yang, J.M. Yu, Z.Q. Xu, S.X. Cui, L. Zhu, M. Tan, X. Jiang, Z.Y. Fang, J. Med. Virol. 80, 1997–2004 (2008)
A. Kroneman, H. Vennema, K. Deforche, H.V. Avoort, S. Peñaranda, M.S. Oberste, J. Vinjé, M. Koopmans, J. Clin. Virol. 51, 121–125 (2011)
B. Lopman, H. Vennema, E. Kohli, P. Pothier, A. Sanchez, A. Negredo, J. Buesa, E. Schreier, M. Reacher, D. Brown, J. Gray, M. Iturriza, C. Gallimore, B. Bottiger, K.O. Hedlund, M. Torvén, C.H. von Bonsdorff, L. Maunula, M. Poljsak-Prijatelj, J. Zimsek, G. Reuter, G. Szücs, B. Melegh, L. Svennson, Y. van Duijnhoven, M. Koopmans, Lancet 363, 682–688 (2004)
R.A. Bull, E.T. Tu, C.J. McIver, W.D. Rawlinson, P.A. White, J. Clin. Microbiol. 44, 327–333 (2006)
T.G. Phan, T. Kuroiwa, K. Kaneshi, Y. Ueda, S. Nakaya, S. Nishimura, A. Yamamoto, K. Sugita, T. Nishimura, F. Yagyu, S. Okitsu, W.E. Müller, N. Maneekarn, H. Ushijima, J. Med. Virol. 78, 971–978 (2006)
C.I. Gallimore, M. Iturriza-Gomara, J. Xerry, J. Adigwe, J.J. Gray, Arch. Virol. 152, 1295–1303 (2007)
D.J. Allen, J.J. Gray, C.I. Gallimore, J. Xerry, M. Iturriza-Gómara, PLoS One 3, e1485 (2008)
K. Sdiri-Loulizi, K. Ambert-Balay, H. Gharbi-Khelifi, N. Sakly, M. Hassine, S. Chouchane, M.N. Guediche, P. Pothier, M. Aouni, J. Clin. Microbiol. 47, 421–429 (2009)
D.P. Zheng, M.A. Widdowson, R.I. Glass, J. Vinjé, J. Clin. Microbiol. 48, 168–177 (2010)
D.S. Cheon, J. Lee, K. Lee, S. Lee, K. Park, J. Ahn, Y. Jee, J. Yoon, H. Cho, J. Med. Virol. 73, 439–442 (2004)
G. Lee, C. Lee, J. Microbiol. 46, 319–324 (2008)
L. Li, Y. He, H. Yang, J. Zhu, X. Xu, J. Dong, Y. Zhu, Q. Jin, J. Clin. Microbiol. 43, 3835–3839 (2005)
K. Katayama, H. Shirato-Horikoshi, S. Kojima, T. Kageyama, T. Oka, F. Hoshino, S. Fukushi, M. Shinohara, K. Uchida, Y. Suzuki, T. Gojobori, N. Takeda, Virology 299, 225–239 (2002)
T. Kageyama, M. Shinohara, K. Uchida, S. Fukushi, F.B. Hoshino, S. Kojima, R. Takai, T. Oka, N. Takeda, K. Katayama, J. Clin. Microbiol. 42, 2988–2995 (2004)
N. Jothikumar, J.A. Lowther, K. Henshilwood, D.N. Lees, V.R. Hill, J. Vinjé, Appl. Environ. Microbiol. 71, 1870–1875 (2005)
S. Kojima, T. Kageyama, S. Fukushi, F.B. Hoshino, M. Shinohara, K. Uchida, K. Natori, N. Takeda, K. Katayama, J. Virol. Methods 100, 107–114 (2002)
Acknowledgments
We thank Dr. J.I. Lee (Seoul Metropolitan Research Institute of Public Health & Environment, Korea) for kindly providing the stool samples and co-workers (Mogam Biotechnology Research Institute, Korea) for discussions that helped us to improve the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Gyu-Cheol Lee and Gyoo Seung Jung contributed equally to this study.
Rights and permissions
About this article
Cite this article
Lee, GC., Jung, G.S. & Lee, C.H. Complete genomic sequence analysis of norovirus isolated from South Korea. Virus Genes 45, 225–236 (2012). https://doi.org/10.1007/s11262-012-0779-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-012-0779-9