
Abstract
RD-114 virus is a replication-competent feline endogenous retrovirus. RD-114 virus contaminates several feline and canine live attenuated vaccines and the issue of contamination of RD-114 virus in vaccines should be solved. To date, three infectious molecular clones (pSc3c, pCRT1, and pRD-UCL) have been reported. In this study, we sequenced the entire nucleotide sequence of pRD-UCL and compared the nucleotide sequences of the three infectious molecular clones. As a result, these three infectious clones were nearly identical with each other in gag-pol and env coding regions. These data support the notion that the active locus of infectious RD-114 virus is single in the feline genome. The length of long terminal repeat (LTR) of pCRT1 was 47 bp shorter than those of pSc3c and pRD-UCL. The 47-bp sequence named direct repeat A (DR-A) was duplicated in the U3 region in pSc3c and pRD-UCL. Although several potential enhancer binding sites are present in the DR-A, there was no significant difference in promoter activities between the LTRs of pRD-UCL and pCRT1 in two human cell lines. We also analyzed the splicing pattern of the RD-114 virus by reverse transcription-polymerase chain reaction and confirmed that RD-114 virus is a simple retrovirus. The data presented here will provide basic information about RD-114 virus to solve the contamination issue in live attenuated vaccines.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
RD-114 virus is a replication-competent feline endogenous retrovirus [1, 2]. Recently, we and others reported that RD-114 virus contaminated several feline and canine live attenuated vaccines [3–6]. RD-114 virus can infect and grow efficiently in canine primary cells as well as established cell lines [7]; therefore, the issue of contamination of RD-114 virus in canine vaccines might be problematic and should be solved.
In the feline genome, there are several copies of RD-114 virus-related sequences [1, 2]. RD-114 virus is a chimeric virus consisting of a gammaretrovirus in gag-pol region and a betaretrovirus in env region [8]. The gag-pol region of RD-114 virus is highly homologous to another feline endogenous gammaretrovirus, termed Felis catus endogenous retrovirus (FcEV). To the contrary, the env region of RD-114 virus is highly homologous to baboon endogenous retrovirus (BaEV). Therefore, RD-114 virus is considered to have arisen by recombination between FcEV and BaEV in cats in the past [8]. The recombination breakpoints reside in the intergenic region between pol and env genes and in the 3′ untranslated region (UTR) downstream of the env gene [8]. To date, three infectious molecular clones have been reported. In this study, we compared the nucleotide sequences and promoter activities of the infectious molecular clones and examined the splicing pattern of RD-114 virus.
The first molecular infectious clone of RD-114 virus, termed pSc3c, was cloned by Reeves and O’Brien [1]. This clone is considered to be derived from the DNA of human rhabdomyosarcoma chronically infected with the original RD-114 virus reported in 1972 [9]. Recently, we constructed two additional infectious molecular clones, termed pCRT1 and pRD-UCL. The former is derived from RD-114 virus produced from Crandell-Rees feline kidney cells (a feline renal cell line; ATCC CCL-94) [5]. To construct pRD-UCL, formerly named as pTE-RD114, genomic DNA was isolated from TE671 cells (a human rhabdomyosarcoma cell line; ATCC CRL-8805) chronically infected with RD-114 virus [13], kindly provided by Dr. Takeuchi (University College London, London, UK). This strain had been used in our previous studies as a reference RD-114 virus [10–12]. pRD-UCL was previously used to analyze the involvement of the late domain of RD-114 virus Gag protein in viral budding [14]. The cloning strategy of pRD-UCL was the same with that of pCRT1 [5]. Infectivity of the clone was confirmed by the LacZ marker rescue assay [10].
So far, the entire nucleotide sequences of pSc3c and pCRT1 have been deposited in GenBank (Table 1). The nucleotide sequence of pSc3c has been deposited by three independent research groups including us. The env sequence in an Env expression plasmid of RD-114 virus, termed pRDF (GenBank accession number: X87829) [15], is different (3 bp-deletion and 10 bp-substitutions) from the others although the env region was derived from the clone pSc3c. To confirm the sequence integrity of the env sequence of the clone pRDF, we amplified the env region from the genomic DNA isolated from TELCeB6 cells stably transduced with pRDF [15, 16] by polymerase chain reaction (PCR) using PrimeSTAR GXL DNA polymerase (Takara, Ohtsu, Shiga, Japan) according to the manufacturer’s instruction and determined the nucleotide sequence of the amplicon by direct sequencing. Direct sequencing was performed by a commercial DNA sequencing service (FASMAC Co., Ltd., Kanagawa, Japan). The nucleotide sequence of the amplicon was identical to that of the env region of pSc3c reported by Ghani et al. [17] (GenBank accession number: NC_009889) and us (GenBank accession number: AB705392). We also determined the entire sequence of pRD-UCL (GenBank accession number: AB705393) and compared it with the other clones. Comparing with the sequence of pCRT1, there is one nucleotide substitution causing one amino acid substation in the env region of the pRD-UCL (Fig. 1). In addition, there are four nucleotide substitutions causing three amino acid substitutions in the gag-pol region (Fig. 1). A successive 3-base-pair (bp) deletion (1-amino-acid deletion) in pSc3c (NC_0098889) was not found in pCRT1 and pRD-UCL. Because this deletion was not found in the nucleotide sequence of pSc3c that we had sequenced (GenBank accession number: AB705392), we think that this deletion is due to a sequencing error or occurred during the preparation of the plasmid in Escherichia coli.

Comparison of nucleotide sequences of the gag-pol and env genes of RD-114 virus clones, pSc3c, pCRT1, and pRD-UCL. The first nucleotide for the initiation codons of the Gag-Pol precursor polyprotein and the Env protein of pCRT1 are defined as nucleotide position +1. Small letters and dashes indicate nucleotides and deletions, respectively. Capital letters in parentheses indicate amino acids
Previously, we reported that the length of long terminal repeat (LTR) of pCRT1 is 47 bp shorter than that of pSc3c. Sequencing analysis revealed that the sequence of LTR of pRD-UCL was identical to that of pSc3c. These data strongly suggest that the RD-114 virus of the pRD-UCL was derived from the original isolate [9]. The 47-bp sequence is repeated in the U3 region of LTR in pSc3c and pRD-UCL and called as direct repeat A (DR-A) (Fig. 2). The DR-A is followed by another direct repeat, named DR-B [18]. Possibly, the DR-A was duplicated during the replication in human or feline cells in vitro [5]. By means of TFSEARCH [19], several transcriptional factors which could bind to the U3 region of LTR were found with high scores (more than 95 points) (Fig. 2). In the DR-A, potential enhancer-binding sites for alcohol dehydrogenase regulator 1, CdxA, heat shock transcriptional factor, and NF-kB were present (Fig. 2).

Comparison of the U3 region of the LTR of RD-114 virus clones, pSc3c, pCRT1, and pRD-UCL. DR-A direct repeat A, DR-B direct repeat B, CAAT CAAT box, TATA TATA box, TSS Transcription start site, poly A poly A signal, ADR1 alcohol dehydrogenase regulator 1, and HSF heat shock transcriptional factor
To know the significance of the duplication of DR-A in the viral promoter activity, we constructed reporter plasmids, termed pGL-UCL-LTR, pGL-UCL-LTR(+), pGL-CRT1-LTR, and pGL-CRT1-LTR(+), in the backbone of pGL3-basic reporter plasmid containing the firefly luciferase gene as a reporter (Promega, Madison, WI, USA), as shown in Fig. 3. The reporter plasmids were constructed using In-Fusion® HD Cloning Kit (Clontech, Mountain View, CA, USA). The detailed procedure for the plasmid construction is available on request. In pGL-UCL-LTR and pGL-CRT1-LTR, the LTR sequences of pRD-UCL and pCRT1, respectively, were placed upstream of the luciferase gene. In pGL-UCL-LTR(+) and pGL-CRT1-LTR(+), each LTR sequence followed by each of the 5′ UTR was placed upstream of the reporter gene. As an internal control, we used pRL-TK vector (Promega) containing Renilla luciferase reporter gene under the control of herpes simplex virus 1 thymidine kinase promoter. Human embryonic kidney (HEK) 293T (ATCC CRL-11268) and TE671 cells subcultured in 12-multiwell plates for 24 h were transfected with each reporter plasmid (50 ng) and pRL-TK (5 ng) using Lipofectamine 2000 (Invitrogen, Carlsbud, CA, USA) according to the manufacturer’s instruction. Four hour after transfection, the medium was replaced with the growth medium. Two days after transfection, the luciferase activities were measured using a dual-luciferase reporter assay system (Promega) with a Lumat LB9507 (Berthold Technologies, Calmbacher, Bad Wildbad, Germany) according to the manufactures’ instructions. As shown in Fig. 3, we could not find any differences between promoter activities of pGL-UCL-LTR and pGL-CRT1-LTR in both HEK293T and TE671 cells. Similarly, we could not find any differences between promoter activities of pGL-UCL-LTR(+) and pGL-CRT1-LTR(+). These data suggest that the repeat of DR-A does not affect promoter activities in these cells.

The LTR promoter activities of pRD-UCL and pRD-CRT1. Each reporter plasmid (50 ng) was cotransfected with pRL-TK (5 ng), a control plasmid expressing Renilla luciferase, into HEK293T cells or TE671 cells. Two days after transfection, the promoter activities were measured by dual-luciferase reporter assay. The firefly luciferase activity of each reporter plasmid relative to Renilla luciferase activity is shown. Assays were performed in triplicate and all results are shown as the mean ± SEM. Luc luciferase
To elucidate the splicing pattern of RD-114 virus, we carried out the reverse transcription (RT)-PCR. HEK293T cells were transfected with the RD-114 infectious molecular clone, pCRT1. One day after transfection, total RNA was extracted from the cells using a RNeasy Plus Mini Kit (QIAGEN, Valencia, CA, USA) according to the manufacturer’s instructions. RT was performed with an oligo-dT20 primer using SuperScript III (Invitrogen) according to the manufacturer’s instructions. PCR was performed with a primer pair U5-Fwd (5′-TAGGAGTTTGGCTCCTAAC-3′; nucleotide position 416-434) and U3-Rev (5′-GTGGGGGGTTCTGACTTCTC-3′; nucleotide position 7,901–7,882) (positions are shown in Fig. 4b). PCR conditions were as follows: the reaction mixture (50 μl) consisted of 1 μl of cDNA template, 10 μl of 5× buffer containing 20 mM MgCl2 (PrimeSTAR GXL buffer; TaKaRa), 1 μl PrimeSTAR GXL DNA polymerase (TaKaRa), 4 μl of 2.5 mM deoxynucleotide triphosphates, 1 μl of each primer (10 μM), and 31 μl distilled water. The amplification conditions were 98 °C for 2 min, followed by 35 cycles of amplification consisting of denaturation at 98 °C for 10 s, annealing at 55 °C for 15 s, extension at 68 °C for 1 min 25 s, and a final extension at 68 °C for 10 min. PCR was carried out in 200-μl thin-walled PCR tubes using a thermalcycler C1000 Thermal Cycler (BIO-RAD, Hercules, CA, USA). As a control, we also isolated genomic DNA from HEK293T cells infected with RD-114 virus clone CRT1 using a QIAamp DNA Blood Kit (QIAGEN) and conducted PCR as the above condition. The amplicons were cloned into pCR4Blunt-TOPO (Invitrogen) and sequenced by a commercial DNA sequencing service (FASMAC Co., Ltd.).

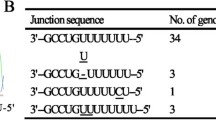
a Schematic structure of RD-114 virus genome. The nucleotide positions of pCRT1 (Accession number AB559882) is shown. b Positions of primers U5-Fwd and U3-Rev used for RT-PCR analysis. c RNA transcripts of RD-114 virus. The nucleotide positions of pCRT1 are also shown. The sequences of splice junctions of RD-114 virus, feline leukemia virus subgroup A (FeLV-A), murine leukemia virus (MLV) are shown under the spliced transcripts. SD splice donor, SA splice acceptor. d RT-PCR. RNA was isolated from HEK293T cells infected with RD-114 virus (Lanes 1 and 3) or mock-infected HEK293T cells (Lanes 2 and 4). DNA was also isolated from HEK293T cells infected with RD-114 virus (Lane 5) or mock-infected HEK293 cells (Lane 6). RT-PCR was carried out using a primer pair U5-Fwd and U3-Rev (Lanes 1 and 2). PCR was carried out from RNA (Lanes 3 and 4) or DNA (Lanes 5 and 6) using a primer pair U5-Fwd and U3-Rev (Lanes 3–6). Lane M 1 kb ladder DNA marker (New England BioLabs, Ipswich, MA, USA)
By RT-PCR, using a primer pair U5-Fwd and U3-Rev, 7.5 and 2.2 kb bands were detected, respectively (Fig. 4d). We could not detect any bands by PCR without RT using RNA as template, indicating that there was no contamination of genomic DNA (Fig. 4d). Sequencing analysis revealed that the 7.5-kb band was a full length RD-114 viral RNA transcript. On the other hand, the 2.2-kb band was a spliced transcript for env mRNA. The splice junction was the same with a previous report by Kharaytonchyk and Pedersen [20], and fairly conserved with other gammaretroviruses, such as feline leukemia virus and murine leukemia virus (Fig. 4c) [21]. We could not find any additional spliced transcripts other than the two transcripts.
In this study, we compared three infectious molecular clones of RD-114 virus. These three infectious clones are nearly identical with each other in coding regions. These data support the notion that the active locus of infectious RD-114 virus is single in the feline genome [2]. Furthermore, we analyzed the splicing pattern of the virus and confirmed that RD-114 virus is a simple retrovirus. The data presented here will provide basic information about RD-114 virus so as to solve the contamination issue in live attenuated vaccines.
References
R.H. Reeves, S.J. O’Brien, J. Virol. 52, 164–171 (1984)
R.H. Reeves, W.G. Nash, S.J. O’Brien, J. Virol. 56, 303–306 (1985)
T. Miyazawa, Biologicals 38, 371–376 (2010)
T. Miyazawa, R. Yoshikawa, M. Golder, M. Okada, H. Stewart, M. Palmarini, J. Virol. 84, 3690–3694 (2010)
R. Yoshikawa, E. Sato, T. Igarashi, T. Miyazawa, J. Clin. Microbiol. 48, 3366–3369 (2010)
R. Narushima, T. Shimazaki, T. Takahashi, Biologicals 39, 89–93 (2011)
R. Yoshikawa, J. Yasuda, T. Kobayashi, T. Miyazawa, J. Gen. Virol. 93, 603–607 (2012)
A.C. Van der Kuyl, J.T. Dekker, J. Goudsmit, J. Virol. 73, 7994–8002 (1999)
R.M. McAllister, M. Nicolson, M.B. Gardner, R.W. Rongey, S. Rasheed, P.S. Sarma, R.J. Huebner, M. Hatanaka, S. Oroszlan, R.V. Gilden, A. Kabigting, L. Vernon, Nat. New Biol. 235, 3–6 (1972)
S. Sakaguchi, M. Okada, T. Shojima, K. Baba, T. Miyazawa, J. Vet. Med. Sci. 70, 785–790 (2008)
S. Sakaguchi, K. Baba, M. Ishikawa, R. Yoshikawa, T. Shojima, T. Miyazawa, J. Vet. Med. Sci. 70, 1383–1386 (2008)
M. Okada, R. Yoshikawa, T. Shojima, K. Baba, T. Miyazawa, Virus Res. 155, 268–273 (2011)
Y. Takeuchi, F.-L. Cosset, P.J. Lachmann, H. Okada, R.A. Weiss, M.K. Collins, J. Virol. 68, 8001–8007 (1994)
A. Fukuma, M. Abe, S. Urata, R. Yoshikawa, Y. Morikawa, T. Miyazawa, J. Yasuda, Virol. J. 8, 540 (2011)
F.-L. Cosset, Y. Takeuchi, J.-L. Battini, R.A. Weiss, M.K.L. Collins, J. Virol. 69, 7430–7436 (1995)
Y. Takeuchi, C. Patience, S. Magre, R.A. Weiss, P.T. Banerjee, P. Le Tissier, J.P. Stoye, J. Virol. 72, 9986–9991 (1998)
K. Ghani, S. Cottin, P.O. de Campos-Lima, M.C. Caron, M. Caruso, J. Gene Med. 11, 664–669 (2009)
D.A. Spodick, A.K. Ghosh, S. Parimoo, P. Roy-Burman, Virus Res. 9, 263–283 (1988)
T. Heinemeyer, E. Wingender, I. Reuter, H. Hermjakob, A.E. Kel, O.V. Kel, E.V. Ignatieva, E.A. Ananko, O.A. Podkolodnaya, F.A. Kolpakov, N.L. Podkolodny, N.A. Kolchanov, Nucleic Acids Res. 26, 362–367 (1998)
S. Kharytonchyk, F.S. Pedersen, RNA 16, 572–584 (2010)
M.D. Papenhausen, J. Overbaugh, Virology 195, 804–807 (1993)
Acknowledgments
We are grateful to Dr. Yasuhiro Takeuchi (University College London, London, UK) for providing TELCeB6/pRDF cells, TE671 cells, TE671 cells chronically infected with RD-114 virus. We thank Dr. Roger Reeves (Johns Hopkins University, Baltimore, MD, USA) for providing pSc3c. This work was supported by a grant-in-aid from the Bio-oriented Technology Research Advancement Institution of Japan.
Author information
Authors and Affiliations
Corresponding author
Additional information
Sayumi Shimode and Rokusuke Yoshikawa have contributed equally to this study.
Rights and permissions
About this article
Cite this article
Shimode, S., Yoshikawa, R., Hoshino, S. et al. Sequence comparison of three infectious molecular clones of RD-114 virus. Virus Genes 45, 393–397 (2012). https://doi.org/10.1007/s11262-012-0759-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-012-0759-0