
Abstract
Lentiviral vectors modified from human immunodeficiency virus type 1 (HIV-1) offer a promising approach for gene therapy, facilitating transduction of genes into non-dividing cells both in vitro and in vivo. When transducing cytotoxic or anti-HIV genes, however, the vector must avoid self-inhibition by the transgene that can lead to a disruption in production of infectious virions. In this study, we constructed two HIV-1-based lentiviral vectors harboring the mifepristone-inducible gene expression unit in either the forward or the reverse orientation with respect to the direction of viral genomic RNA. The ability of these vectors to transduce cytotoxic and anti-HIV genes was evaluated. When human CD14 was used as a transgene, infectious lentiviral vectors were produced by both forward and reverse vector systems. CD14 expression was efficiently induced in cells transduced by both lentiviral vectors following treatment with mifepristone. However, a higher level of basal transgene expression was observed in the forward vector system in the absence of mifepristone. In contrast, high titers of infectious lentiviral vector containing the cytotoxic vesicular stomatitis virus M gene were successfully generated using the reverse vector, but not the forward vector. In addition, when a VPS4B-dominant negative mutant against HIV-1 budding was cloned into the reverse vector, significant amounts of lentiviral vector were obtained. Subsequent transduction of cells with the VPS4B mutant resulted in approximately 50% inhibition of HIV-1 production only in the presence of mifepristone. Our study thus demonstrates that incorporation of a mifepristone-regulatable gene expression unit in the reverse orientation makes significant advances toward development of a lentiviral vector that allows transduction of harmful genes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A variety of gene-transfer vectors based on RNA and DNA viruses have been developed to deliver foreign genes to target cells in vitro and in vivo [1]. Retroviral vectors derived from gammaretroviruses and lentiviruses have the potential advantage of sustained expression of transgenes in transduced cells, because of their ability to stably integrate viral DNA into the host genome. While gammaretroviruses require cell division to establish infections, lentiviruses including human immunodeficiency virus type 1 (HIV-1) are capable of infecting both dividing and non-dividing cells [1]. Lentivirus-based vector systems thus potentiate long-term gene expression in non-dividing cells such as neurons and hematopoietic stem cells [2, 3].
Lentiviral vectors hold great promise for a gene therapy approach to inherited and acquired diseases such as cancer and acquired immunodeficiency syndrome (AIDS). A potential application of lentiviral vectors would be the transduction of a gene cytotoxic to tumor cells or virus-infected cells resulting in the eradication of these unwanted cells from the body. For the treatment of AIDS, an alternative approach would be to deliver an anti-HIV gene to a population of cells rendering them resistant to HIV infection. However, insertion of toxic or anti-HIV genes into an HIV-based lentiviral vector can create problems for production of the vector itself. Expression of anti-HIV transgenes in vector packaging cells can interfere with production of lentivirus particles, blocking the ability to make lentiviral vector [4]. One strategy to solve this problem is the use of a regulatable system in which the target transgene is kept silent during vector production and expression is subsequently switched on following transduction in the context of a lentiviral vector.
The first generation of regulatable gene expression systems was based on naturally occurring inducible promoters [5]. However, this type of system had limitations due to high levels of “leaky” or basal expression driven by such promoters, modest induction of transgene expression, and pleiotropic activity of the inducer. For these reasons, the last two decades have seen development of chimeric regulatable systems engineered from a number of prokaryotic, eukaryotic, and viral elements designed to enhance specificity and activity of transgene expression [6]. Amongst the reported chimeric regulators is one based on a mutated human progesterone receptor which is unable to bind endogenous hormone but is activated by binding the progesterone antagonist, mifepristone (RU-486) [7, 8]. The chimeric transactivator (regulator) protein of this so-called GeneSwitch system comprises the GAL4 DNA-binding domain from Saccharomyces cerevisiae fused to the ligand-binding domain of a mutant progesterone receptor and the activation domain of the p65 subunit of human NF-κB [8]. In the presence of mifepristone, this transactivator binds to GAL4 activation sequences upstream of the inducible transgene, stimulating transcription of the target gene by more than 200-fold in cultured cells [9]. An advantage of the GeneSwitch system is that the majority of its components are modified human proteins having no impact on cell viability. In addition, although mifepristone has anti-progesterone and -glucocorticoid activities, the concentration needed for ligand-inducible transactivation of the target gene (10−8 to 10−11 M) is much lower than the concentration producing an anti-progesterone effect in humans [10]. Furthermore, use of a mifepristone-inducible (autoinducible) promoter to regulate expression of the chimeric transactivator dramatically reduced basal expression of the transgene in the absence of the inducer, thereby improving the dynamic range of in vivo transgene regulation [9].
Many types of regulatable gene expression systems have been incorporated into lentiviral vectors [11–21]. The most commonly used inducible system is based on the bacterial tetracycline-responsible gene expression system (Tet system) [22]. While representing an important tool for controlling target gene expression, Tet-regulatable systems in the context of lentiviral vectors have shown high basal levels of transgene expression without induction [22]. Such leakiness would be undesirable especially in the production of lentiviral vectors aimed at transducing toxic proteins into target cells.
To generate a viral vector in which transgene expression was tightly controlled, we combined an HIV-1-based lentiviral vector with the GeneSwitch system described above. The mifepristone-inducible lentiviral vector reported here minimized the interference of transgene expression during virus production and permitted efficient transduction of cytotoxic and anti-HIV genes into target cells.
Materials and methods
Cells
GeneSwitch-293 cells (a HEK293-derived human cell line expressing the GeneSwitch protein) were purchased from Invitrogen. GeneSwitch-293 and human 293T cells were maintained in Dulbecco’s modified Eagle medium (D-MEM) containing 10% fetal calf serum (FCS), 100 units/ml of penicillin, and 100 μg/ml of streptomycin (D-MEM/10% FCS).
Construction of lentiviral vector plasmids
The mifepristone-inducible promoter sequence, GAL4/TATA, was amplified by polymerase chain reaction (PCR) from the vector pGene/V5-His5C (Invitrogen) and subcloned into LITMUS28 (New England Biolabs) to create pLITMUS28-GAL4/TATA. A GAL4/TATA fragment was then inserted into a Gateway-compatible lentiviral vector plasmid pYK005C [23] producing the plasmid for the forward vector (fragment positioned in the forward orientation).
To generate a plasmid for the reverse vector, a fragment containing the Gateway cloning cassette, internal ribosome entry site (IRES), and humanized Renilla green fluorescent protein (hrGFP) sequences together with a fragment containing bovine growth factor hormone poly(A) (BGH pA) sequence were isolated from pYK005C and pSwitch (Invitrogen), respectively. These were subcloned into LITMUS28. Then, the GAL4/TATA sequence derived from pLITMUS28-GAL4/TATA was inserted into the subcloning plasmid. To prepare a vector backbone, the internal human elongation factor 1 α subunit promoter of a self-inactivating (SIN) lentiviral vector plasmid, CSII-EF-MCS [24], was replaced with a fragment containing GAL4/TATA, Gateway-cassette, IRES, hrGFP, and BGH pA sequences, thus generating the final plasmid.
Preparation of all Gateway plasmids containing the ccdB gene was carried out using Escherichia Coli (E. Coli) strain DB3.1.
Cloning of transgenes
All transgenes were cloned into the lentiviral vector plasmid via the Gateway cloning system [23]. Entry plasmid clones encoding transgenes were constructed as follows. Human CD14 was amplified by PCR using primers containing the attB1 tail (5′-GGGGACAAGTTTGTACAAAAAAGCAGGCT-3′) at the 5′-end of the forward primer and the attB2 tail (5′-GGGGACCACTTTGTACAAGAAAGCTGGGT-3′) at the 5′-end of the reverse primer [23]. PCR products were subcloned into the entry plasmid, pDONR201 (Invitrogen), using a Gateway BP reaction [23]. Vesicular stomatitis virus matrix protein (VSV M) cDNA fused with a FLAG epitope tag sequence (FLAG-VSV-M) was generated from a VSV M-expressing plasmid, pEGFPN3-M [25]. This involved use of a forward primer containing the attB1 tail and subsequent FLAG tag sequence (5′-ATGGATTACAAGGATGACGACGATAAG-3′) and a reverse primer containing the attB2 tail. The PCR fragment encoding FLAG-VSV-M was subcloned into entry plasmid, pDONR221 (Invitrogen), by Gateway BP reaction. Fragments encoding the VPS4B K180Q mutant (VPS4B-KQ) or firefly luciferase (Luc) were amplified by PCR from VPS4B K180Q-expressing plasmid [26] and pGL3-Basic (Promega), respectively. These fragments were inserted into EcoRI-MluI sites downstream of three FLAG epitope tags in pCMV-SPORT6-3xFLAG, a Gateway-compatible pCMV-SPORT6 (Invitrogen)-derived entry plasmid. After sequence confirmation, individual transgenes in entry plasmids were transferred to lentiviral vector plasmids by Gateway LR reaction.
Lentiviral vector production
293T cells were seeded to appropriate densities 20 h prior to transfection. Infectious lentiviral vectors pseudotyped with VSV G protein were produced by lentiviral vector plasmid, VSV G- and HIV-1 Rev-expressing plasmid (pCMV-VSV-G-RSV-Rev), and HIV-1 Gag-Pol-expressing plasmid (pCAG-HIVgp) via calcium phosphate-mediated transfection, as described previously [27]. Conditioned medium was harvested 48-h post-transfection and concentrated 40-fold by ultracentrifugation at 4°C at 100,000×g for 90 min.
For titration of the lentiviral vectors, GeneSwitch-293 cells were infected with serial dilutions of vector stocks supplemented with 10 nM mifepristone (Invitrogen) 24-h post-infection. Vector titers (transduction units: TU) were determined 48 h after induction by quantitative flow cytometric analysis for hrGFP positive cells.
In the following text, the use of “p” and “v” to prefix nomenclature denotes vector plasmid and infectious lentiviral vector, respectively.
Transduction and induction of transgenes
GeneSwitch-293 cells were seeded in 6-well plates at a density of 1 × 105 cells/well with D-MEM/10% FCS 20 h prior to infection. Cells were then exposed to lentiviral vectors for 24 h at multiplicities of infection (MOI) of 0.1 (for vF-CD14 and vR-CD14) or 5 (for vR-VSV-M). To induce transgene expression, culture medium was replaced with D-MEM/10% FCS containing 10 nM mifepristone and cells were analyzed 48 h (vF-CD14 and vR-CD14 transductions) or 24 h (for vR-VSV-M transduction) later.
Flow cytometric analysis
Transduced cells were incubated with anti-human CD14 mouse monoclonal antibody (61D3, eBioscience) for 20 min at 4°C and then stained with Cy5-conjugated anti-mouse IgG donkey polyclonal antibody (Chemicon International Inc.) for a further 20 min at 4°C. Data were collected using the FACScalibur system (BD Bioscience) and analyzed with WinMDI software.
Western blotting analysis
Cells were lysed in SDS sample buffer (62.5 mM Tris–HCl, pH 6.8, 2% SDS, 5% glycerol, 0.003% bromophenol blue, 0.9% β-mercaptoethanol). Boiled samples were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to Immobilion Transfer Membranes (Millipore). Primary antibodies used were (i) anti-FLAG mouse monoclonal IgG (M2, Sigma) to detect FLAG-VSV-M; (ii) biotinylated anti-FLAG mouse monoclonal IgG (BioM2, Sigma) to detect FLAG-VPS4B-KQ and FLAG-Luc; and (iii) anti-α-tubulin mouse monoclonal IgG (DM1A, Sigma) to detect α-tubulin. Biotinylated anti-mouse IgG (BA-2000, Vector Laboratories) was then used for the detection of FLAG-VSV-M. Proteins were detected using horseradish peroxidase (HRP)-conjugated streptavidin (ZYMED Laboratories, for FLAG-tagged proteins) and HRP-conjugated anti-mouse IgG (Cell Signaling, for α-tubulin) using a Western Lightning Chemiluminescence Reagent Plus (PerkinElmer). Signals were analyzed using a luminescence image analyzer, LAS-3000mini (Fujifilm).
Microscopy analysis
293T cells transfected with pF-VSV-M and pR-VSV-M were examined by light and fluorescent microscopy using an Eclipse TS100 microscope (Nikon) at the point of harvesting lentiviral vector. Images were obtained with a DFC480 digital camera and IM500 image manager software (Leica Microsystems).
To detect FLAG-VSV-M, transduced cells were fixed with 4% paraformaldehyde and incubated with PBS containing 5% normal goat serum and 0.05% Triton X-100 at room temperature for 1 h. This was followed by incubation with anti-FLAG-mouse monoclonal IgG (M2) at 4°C overnight. Samples were then incubated with Alexa Fluor 594-conjugated anti-mouse IgG (Invitrogen) at room temperature for 1 h and nuclei were stained using Hoechst 33342 (Invitrogen). Images were obtained using a CTR 6500 fluorescent microscope and FW4000 software (Leica Microsystems).
Assay for HIV-1 release from VPS4B mutant-transduced cells
GeneSwitch-293 cells were seeded at a density of 1 × 103 cells/well in a 96-well plate 20 h prior to infection and exposed to vR-VPS4B-KQ and vR-Luc at a MOI of 1 for 24 h. Transduced cells were then expanded for 4–5 weeks and reseeded at a density of 4 × 106 cells in a 15-cm diameter dish 20 h prior to induction. A portion of transduced cells was subjected to flow cytometric analysis to monitor hrGFP expression after induction with 10 nM mifepristone. To enrich transduced cells, single hrGFP positive cells were positively selected using the FACSAria cell sorting system (BD Bioscience). Sorted cells were cultured for a further 4–5 weeks and cell clones with the strongest hrGFP signals in respective transductions were used for an assay of HIV-1 release.
For the assay of HIV-1 release, 4 μg of pNL4-3 [28] was transfected to transduced cells with Lipofectamine 2000 (Invitrogen). Four hours post-transfection, culture medium was replaced with D-MEM/10% FCS containing 10 nM mifepristone. Culture supernatant was harvested 24 h later and virus production was monitored by HIV-1 p24CA enzyme-linked immunosorbent assay (ELISA, ZeptoMetrix).
Statistical analysis
Student’s t test was used to determine statistical significance. A P value of <0.05 was considered significant.
Results
Generation of mifepristone-inducible lentiviral vectors
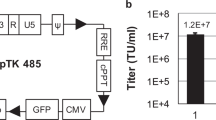
To establish a regulatable viral vector system, we cloned a mifepristone-inducible promoter sequence (as an internal promoter) into a self-inactivating (SIN) lentiviral vector plasmid in which the U3 region of the 5′-long terminal repeat (LTR) was replaced with the cytomegalovirus (CMV) promoter and the enhancer/promoter unit was deleted from the U3 region of the 3′-LTR [23, 27]. The inducible promoter was a hybrid consisting of the yeast GAL4 upstream activating sequences linked to the adenovirus major late E1b TATA box (GAL4/TATA) [7]. Gene expression from the GAL4/TATA promoter was controlled by a chimeric regulatory protein termed GeneSwitch. Binding of mifepristone to GeneSwitch induced a conformational change in the regulator to an active state, resulting in transcription of the gene of interest [7]. Besides an inducible promoter, our lentiviral vectors contained a Gateway cloning system reading frame cassette, facilitating cloning of genes of interest by site-specific recombination-based Gateway technology [23]. In addition to a conventional lentiviral vector containing the GAL4/TATA-Gateway component in the forward orientation (forward vector, Fig. 1a), we also constructed a version of the lentiviral vector plasmid in which the inducible gene expression unit was located in the reverse orientation, with the intention of reducing interference from the CMV promoter (reverse vector, Fig. 1a).

Transduction of the CD14 gene by mifepristone-inducible lentiviral vectors. a Schematic of forward and reverse vectors. In these SIN lentiviral vector plasmids, the U3 region of the 5′-LTR has been replaced with the CMV promoter (CMVP) resulting in Tat-independent transcription. A portion of the U3 region containing the enhancer/promoter unit has been deleted from the 3′-LTR (represented as triangles). The gene of interest (transgene), flanked by attB sites, was subcloned into an entry plasmid and then transferred to the vector plasmid by a Gateway reaction. Note that site-specific recombination between attR sites on the vector plasmid and attL sites on the entry vector gave rise to new attB sites on the final lentiviral vector plasmid. The GAL4/TATA promoter regulated expression of the transgene and IRES-controlled hrGFP gene in the presence of the transactivator protein (GeneSwitch) and mifepristone. Ψ, packaging signal; RRE Rev responsive element; WPRE woodchuck post-regulatory element, BGH pA bovine growth factor hormone poly(A). b Expression of CD14 in vector producer cells. 293T cells were co-transfected with a VSV G/Rev-expressing plasmid, a Gag-Pol-expressing plasmid, and a forward or reverse vector plasmid coding CD14 and hrGFP genes (pF-CD14 and pR-CD14, respectively). Expression of CD14 (y-axis) and hrGFP (x-axis) at the time of vector harvest (48 h after transfection) was analyzed by flow cytometric analysis. Untransfected cells (mock) served as a negative control. The representative results of three independent experiments are shown. c Infectious titers of lentiviral vectors. Vector titer (transducing units [TU]/ml) was determined by quantitative flow cytometric analysis for hrGFP positive cells on transduced GeneSwitch-293 cells in the presence of mifepristone. Values represent the mean ± standard deviation (SD) for triplicate determinations. d Transduction and induction activities of lentiviral vectors. GeneSwitch-293 cells were infected with vF-CD14 or R-CD14 at a MOI of 0.1 (or uninfected, mock) and cultivated in the presence or absence of 10 nM mifepristone (Mif (+) and Mif (−), respectively). Expression of CD14 protein 48 h after induction was determined by flow cytometric analysis. Values represent the mean ± SD of three independent experiments. The P value versus no mifepristone treatment is <0.05 by Student’s t test (*)
To evaluate our gene delivery system, the human CD14 gene was cloned into both forward and reverse vector plasmids (designated pF-CD14 and pR-CD14, respectively). Infectious lentiviral vectors (vF-CD14 and vR-CD14) were produced following co-transfection of VSV G/Rev-expressing plasmid and Gag-Pol-expressing plasmid into 293T cells. Flow cytometric analysis of transfected cells showed that even in the absence of the GeneSwitch protein and mifepristone, expression of CD14 and cistronic hrGFP genes occurred in pF-CD14-transfected producer cells (40.7 ± 3.1%), while such expression was repressed in pR-CD14-transfected cells (1.4 ± 0.4%) (Fig. 1b). This leaky expression of transgenes from the forward vector plasmid in virus producer cells might be due to transcriptional interference arising from the presence of a heterologous promoter in the same orientation (i.e., the CMV promoter), which was required for Tat-independent transcription of the viral RNA genome [27]. Culture supernatant containing the lentiviral vector was harvested and viral titer determined by quantification of the number of hrGFP-positive cells in viral vector-transduced GeneSwitch-293 cells, expressing the GeneSwitch regulatory protein, in the presence of mifepristone [8]. Lentiviral vectors produced by pF-CD14 showed 34.4-fold higher viral titers (vF-CD14, 2.0 ± 0.5 × 106 TU/ml) than viral vectors obtained by pR-CD14 (vR-CD14, 5.8 ± 1.1 × 104 TU/ml) (Fig. 1c). The transduction and induction efficiencies of lentiviral vectors were then assessed following infection of GeneSwitch-293 cells at a MOI of 0.1. As shown in Fig. 1d, a low level of CD14 expression in transduced cells was observed in both vF-CD14- and vR-CD14-infected cells in the absence of mifepristone. However, basal gene expression in vF-CD14-infected cells (16.5 ± 1.7%) was higher than that recorded in vR-CD14-infected cells (6.3 ± 2.2%), indicating that transgene expression from the reverse orientation vector was tightly controlled in both transduced cells and virus producer cells (Fig. 1b, d). In the presence of mifepristone, expression of CD14 increased significantly, with the percentage of CD14-positive cells rising to 78.6 ± 0.6% (vF-CD14) and 44.3 ± 9.9% (vR-CD14) (Fig. 1d). The regulation factor (ratio of maximal induced expression to basal expression levels in the absence of the inducer [29]) of vR-CD14 was higher than that of vF-CD14 (7.0-fold vs. 4.7-fold). These results demonstrated that our mifepristone-inducible lentiviral vectors functioned as an efficient gene-delivery system with a tight on–off switch for regulating transgene expression.
Transduction of a cytotoxic gene by the mifepristone-inducible lentiviral vector
In gene therapies for cancer patients, delivery of a cytotoxic gene by lentiviral vector is one strategy for destroying tumor cells. However, expression of a toxic gene in a lentiviral vector genome would hamper vector production resulting in a reduction in titer. Expression of such a cytotoxic gene thus needs to be blocked during the process of vector production. The lentiviral vector described here, containing a mifepristone-inducible gene expression unit in the reverse orientation, shows great promise as a carrier for a cytotoxic transgene with CD14 expression tightly repressed in pR-CD14-transfected producer cells (Fig. 1b). We examined this system further to ascertain if it could produce infectious lentiviral vector containing a cytotoxic gene and transduce that gene into target cells. Forward and reverse vector plasmids containing FLAG tag-fused VSV matrix protein (FLAG-VSV-M) were constructed (pF-VSV-M and pR-VSV-M, Fig. 2a) and transfected into 293T cells to produce VSV G-pseudotyped infectious vector. VSV M was chosen as it inhibits nuclear export of cellular RNA by interacting with nucleoporin. This protein is responsible for most of the cytopathic effects observed in VSV-infected cells [30]. Analysis of the level of FLAG-VSV-M in virus producer cells showed that, even in the absence of inducers, transgene expression was activated in pF-VSV-M-transfected cells, while expression was undetectable in pR-VSV-M-transfected cells (Fig. 2b). As predicted by Western blotting analysis, activated expression of the downstream hrGFP cistron, together with the rounded phenotype of infected cells (a hallmark of VSV infection in cell culture [30]), was observed in pF-VSV-M-transfected cells (Fig. 2c). In contrast, pR-VSV-M-transfected cells exhibited neither hrGFP expression nor cytopathic effect, demonstrating the ability of the reverse vector to suppress the cytotoxic gene in virus producer cells (Fig. 2c). Importantly, the ability of pR-VSV-M to suppress VSV M expression was reflected in the production of infectious lentiviral vector from transfected cells. Transfection with pR-VSV-M yielded lentiviral vector (vR-VSV-M) with titers of 1.5 ± 0.5 × 104 TU/ml, while no infectious vector could be obtained by transfection with pF-VSV-M (Fig. 2d). The ability of vR-VSV-M to transduce the transgene was then analyzed by infection of GeneSwitch-293 cells. Immunostaining analysis indicated that FLAG-VSV-M expression was induced in response to mifepristone in transduced cells (Fig. 2e) and the regulation factor (i.e., inducibility) was 11.8-fold (FLAG positive cells per Hoechst 33342 positive cells, 4.6 ± 3.2% without mifepristone treatment versus 54.5 ± 20.6% with mifepristone treatment (Fig. 2f). These data demonstrated that our reverse vector system is suitable for the transduction of a harmful gene.

Effective transduction of VSV M by the reverse vector. a Forward and reverse vectors containing FLAG-tagged VSV M (FLAG-VSV-M) as a transgene (pF-VSV-M and pR-VSV-M, respectively). b Expression of FLAG-VSV-M in vector producer cells. 293T cells were co-transfected with a VSV G/Rev-expressing plasmid, a packaging plasmid, and pF-VSV-M or pR-VSV-M to produce lentiviral vectors. Cell extracts were prepared 48 h after transfection and subjected to Western blotting (WB) analysis using anti-FLAG (upper panel) and anti-α-tubulin (lower panel) antibodies. c Leaky expression of VSV M caused the death of producer cells. Fluorescent (upper panels) and light (lower panels) microscopic analysis of producer cells was undertaken at the time of vector harvest. Scale bar, 40 μm. d Titers of lentiviral vectors bearing the VSV M gene. TU was determined by hrGFP positive cells in GeneSwitch-293 cells in the presence of mifepristone. The expression of hrGFP in vF-VSV-M-infected cells was below the detection limit (not detected, N.D.). Value of vR-VSV-M represents the mean ± SD of three independent experiments. e Inducible expression of FLAG-VSV-M in vR-VSV-M-transduced cells. GeneSwitch-293 cells were infected with vR-VSV-M at a MOI of 5 and fixed with paraformaldehyde 24 h after induction. Expression of FLAG-VSV-M was examined as described in materials and methods. Hoechst 33342 indicates nucleus. Mif (−), without mifepristone treatment; Mif (+), with 10 nM mifepristone treatment; scale bar, 100 μm. f Quantification of inducibility. FLAG-VSV-M positive cells observed by immunofluorescence analysis were counted in a randomly selected visual field. Data are expressed as the percentage of FLAG+ cells in Hoechst 33342+ cells (mean ± SD of four independent transductions). The P value versus no mifepristone treatment is <0.05 by Student’s t test (*)
Inhibition of HIV-1 release by mifepristone-inducible lentiviral vector
To evaluate whether the reverse vector could overcome self-inhibition caused by an anti-HIV transgene in producer cells, a dominant negative mutant of human VPS4B was selected as a transgene. VPS4B is one of two known isoforms of AAA-ATPase VPS4 and is thought to catalytically remove endosomal sorting complex required for transport (ESCRT) complex III proteins from the plasma membrane, resulting in budding of HIV-1 from infected cells [26]. The ATPase activity of VPS4B is essential for HIV-1 budding with dominant negative mutants of VPS4 (such as K180Q, where the residues required for ATP binding have been mutated), previously reported to block HIV-1 release and infectivity [26]. We inserted a FLAG tag-fused VPS4B K180Q mutant (FLAG-VPS4B-KQ) into both the forward and the reverse vector plasmids and constructed vector plasmids encoding FLAG tag-fused luciferase (FLAG-Luc) as control vectors (Fig. 3a). When these vector plasmids were used to produce lentiviral vector, expression of the transgenes (i.e., FLAG-VPS4B-KQ or FLAG-Luc) was observed in producer cells transfected with forward vector plasmids, but not in cells with reverse vector plasmids (Fig. 3b). While transfection with pR-VPS4B-KQ yielded infectious lentiviral vector, vR-VPS4B-KQ, transfection with pF-VPS4B-KQ did not generate infectious vector, indicating that leaky expression of the VPS4 dominant negative mutant from the forward vector plasmid self-inhibited lentiviral vector production (Fig. 3c). Titers of vR-VPS4B-KQ (8.7 ± 2.4 × 104 TU/ml) were comparable to those of lentiviral vector obtained by transfection with pR-Luc (vR-Luc, 5.9 ± 4.1 × 104 TU/ml), indicating that titers of lentiviral vectors containing the gene expression unit in a reverse orientation were not affected by the anti-HIV transgene (Fig. 3c).

Inhibition of HIV-1 release by a reverse vector encoding VPS4B dominant negative mutant. a Schematic representation of forward and reverse vectors bearing FLAG-tagged VPS4B K180Q (FLAG-VPS4B-KQ) or luciferase (FLAG-Luc). b Expression of transgenes in vector producer cells. The lentiviral vectors were produced as described in “Materials and methods”. Producer cells were collected at the time of virus harvest and subjected to Western blotting (WB) analysis using anti-FLAG (upper panels) and anti-α-tubulin (lower panels) antibodies. c Titers of lentiviral vectors. N.D. means below the detection limit of flow cytometric analysis for hrGFP expression. d Induction of FLAG-VPS4B-KQ and FLAG-Luc expressions by reverse vectors. GeneSwitch-293 cells were infected with vR-VPS4B-KQ or vR-Luc at a MOI of 1 and cultivated in the absence of mifepristone. After sorting of cell clones that were capable of expressing hrGFP, the inducibility of FLAG-VPS4B-KQ and FLAG-luciferase expression was analyzed by western blotting. Mif (−) and Mif (+) indicate before and after induction with mifepristone, respectively. e Analysis of HIV-1 production in transduced cells. GeneSwitch-293 cell lines transduced with either vR-VPS4B-KQ or vR-Luc (and untransduced cells, mock) were transiently transfected with pNL4-3. The amount of HIV-1 virions present in the culture supernatant was measured by p24CA ELISA. The efficiency of HIV-1 release in the presence of 10 nM mifepristone is shown as a percentage of the value in the absence of mifepristone (mean ± SD of three independent transfections). The P value versus no mifepristone treatment is <0.05 by Student’s t test (*)
The ability of our lentiviral vectors to transduce the VPS4B dominant negative mutant for inhibition of HIV-1 release was then tested. GeneSwitch-293 cells were infected with vR-VPS4B-KQ and vR-Luc and sorted for hrGFP positive cells. When transduced cells were cultured with mifepristone, induction of transgene expression was seen (Fig. 3d). The inhibitory effect of transgenes on HIV-1 release from transduced cells was examined by transiently transfecting a plasmid DNA producing infectious HIV-1 virions (pNL4-3). In the presence of mifepristone, levels of HIV-1 production from vR-VPS4B-KQ-transduced cells dropped to 53.9 ± 8.5%, approximately half that seen in the absence of mifepristone treatment. However, no significant inhibitory effects on HIV-1 release were observed in vR-Luc or mock-transduced cells (112.3 ± 9.0% and 89.7 ± 3.1%, respectively). This indicated that induced expression of dominant negative mutant VPS4B proteins in transduced cells accounted for the observed inhibition of HIV-1 release. These results demonstrate the utility of our reverse vector to transduce an anti-HIV gene that functionally suppresses HIV-1 release in target cells.
Discussion
The basic principle of current gene therapy is to deliver genetic material to a population of cells in the body, thereby preventing a disease or improving the clinical status of a patient. Although, a key factor in successfully implementing gene therapy is the development of effective vector systems, a number of issues need to be addressed to apply them in a clinical setting. In terms of viral vector systems, one of the major problems is that insertion of cytotoxic or antiviral transgenes adversely affects viral titers during vector production. In this study, we incorporated a mifepristone-inducible gene expression unit into HIV-1-based lentiviral vectors to solve the problem of vector self-inhibition.
Previous studies have reported the delivery of various anti-HIV genes by HIV-1-based vectors in vitro and in vivo [31–36]. Some of the transgenes used in these studies target HIV-1 RNA sequences either directly or indirectly, aiming to inhibit transcription, nuclear translocation, or translation of viral RNA [32–36]. In these types of approach, the problem of self-inhibition can be solved by modifying the nucleotide sequence of a lentiviral vector such that the function of the vector RNA does not interfere with the anti-HIV transgene in producer cells. However, if the transgene targets a fundamental process of the HIV-1 life cycle, such as virion formation, another strategy to avoid self-inhibition is to express the transgene in a regulated manner such that its expression is blocked in producer cells and induced in target cells. This kind of approach would be of value in the transduction of a harmful gene into target cells. The data presented here demonstrate that a lentiviral vector bearing a regulatable gene expression unit is indeed capable of transducing cytotoxic (VSV M) and anti-HIV (VPS4B K180Q) genes into target cells without significant decrease in vector titer (Figs. 2, 3). In addition, induction of anti-HIV genes in transduced cells resulted in approximately 50% inhibition of HIV-1 release (Fig. 3e).
Expression of VPS4B-KQ mutant by transfection has been reported to inhibit HIV-1 release >100-fold [26]; although, the VPS4B-KQ expression induced by our mifepristone-regulatable system produced about 2-fold reduction in HIV-1 production (Fig. 3e). When we looked at the IRES-controlled hrGFP expression in mifepristone-induced cells that had been transduced by vR-VPS4B-KQ or vR-Luc and sorted, hrGFP expressions were only observed in 10.5% (vR-VPS4B-KQ) or 12.2% (vR-Luc) of the cells (data not shown). We speculate that uninducible population of cells was still permissive to HIV-1 production and thus lead to the observed 50% inhibition in the vR-VPS4B-KQ-transduced cells. During expansion of these transduced cells after cell sorting it is possible that some (e.g., gene shut-off) lost their ability to be induced by mifopristone. Besides, the cell sorting step might lead to this issue of lost inducibility. Therefore improving the way to enrich transduced cells should help to alleviate this problem.
To achieve tight regulation of transgene expression, enabling production of infectious vectors, it was necessary to place the mifepristone-inducible gene expression unit in the reverse orientation in the context of the lentiviral vector. Sirin and Park [12] tested the forward and the reverse orientations of a mifepristone-inducible gene expression unit in HIV-1-based lentiviral vectors and reported basal levels of transgene expression that were higher in lentiviral vectors bearing the expression cassette in the reverse orientation than those containing it in the forward orientation. This was in contrast to the findings presented here, where basal expression of the CD14 transgene in reverse vector-infected cells appeared to be lower than that in forward vector-infected cells (Fig. 1d). Similarly to the Sirin and Park [12] study, we used an HIV-1-based SIN vector in which the woodchuck post-regulatory element (WPRE) was inserted into the 3′-untranslated region of the viral genome (Fig. 1a). WPRE has been reported to increase the stability of RNA transcripts, thereby enhancing transgene expression from retroviral and lentiviral vectors [37]. Interestingly, WPRE functions only when placed in the sense orientation of a transgene and antisense WPRE actually shows an inhibitory effect on transgene expression [37]. In our reverse vector, WPRE was positioned in the opposite orientation to the inducible gene expression unit (Fig. 1a), while the vector designed by Sirin and Park [12] contained WPRE in same orientation as the expression unit. Orientation-dependent elements such as WPRE can thus enhance basal expression of a transgene in both producer and transduced cells. In addition to WPRE, the SIN vector used in our study contained a hybrid 5′-LTR in which the U3 region was replaced with the CMV promoter [27]. We speculate that, in the context of our forward vector, these cis-acting sequences should increase background activity of the mifepristone-regulatable gene expression unit without induction, leading to leaky expression of cytotoxic/anti-HIV genes in producer cells and significant loss of vector titers (Figs. 2d, 3c).
One general drawback of regulatable gene expression systems, including the Tet and mifepristone systems, is that they necessitate delivery of two expression units into a target cell; one to express the transactivator and the other to express the transgene in response to the activator. To exclude differences in experimental conditions due to differing levels of transactivator expression, a cell line stably expressing the GeneSwitch transactivator was used as a target cell in this study. While Sirin and Park [12] also described a two-lentiviral vector system in which GeneSwitch and inducible gene expression units were cloned into separate vectors, this type of binary approach would produce populations of singly transduced cells with either transactivator or transgene, resulting in low inducibility. Single-lentiviral vectors bearing the entire regulatable unit have been developed in Tet systems [11, 38, 39]. This single-vector approach would be an attractive option for the mifepristone-regulatable system, bypassing the need for co-transduction of target cells with high amounts of virus. However, RNA virus-based vectors, such as lentiviral vectors, are limited in their cloning capacity for larger genes. Theoretically, lentiviral vectors can accommodate 7–7.5 kb of foreign DNA [1], yet this packaging capacity will be decreased by the insertion of additional regulatory sequences. Improvements to the mifepristone system that would allow incorporation of both transactivator and inducible units into a single-lentiviral vector would be necessary to design a more versatile vector.
The mifepristone-regulatable gene expression system reported here has a number of potential advantages that suit it to gene therapy applications in humans. First, the majority of the system consists of modified human proteins with no impact on cell viability. Second, the induction response is specific and rapid. Third, mifepristone is orally effective and the dose required for induction is within the range acceptable for clinical use [10]. Importantly, mifepristone has been approved by the Food and Drug Administration (FDA) for use in humans. Although, no gene regulatory system has yet been approved by the FDA for clinical use, lentiviral vectors in conjunction with a mifepristone-regulatable gene expression system are a promising step toward achieving successful gene therapy.
References
I.M. Verma, N. Somia, Nature 389, 239–242 (1997)
L. Naldini, U. Blomer, P. Gallay, D. Ory, R. Mulligan, F.H. Gage, I.M. Verma, D. Trono, Science 272, 263–267 (1996)
H. Miyoshi, K.A. Smith, D.E. Mosier, I.M. Verma, B.E. Torbett, Science 283, 682–686 (1999)
J.A. Taylor, L. Vojtech, I. Bahner, D.B. Kohn, D.V. Laer, D.W. Russell, R.E. Richard, Mol. Ther. 16, 46–51 (2008)
M. Fussenegger, Biotechnol. Prog. 17, 1–51 (2001)
S. Agha-Mohammadi, M.T. Lotze, J. Clin. Invest. 105, 1177–1183 (2000)
Y. Wang, B.W. O’Malley Jr., S.Y. Tsai, B.W. O’Malley, Proc. Natl Acad. Sci. USA 91, 8180–8184 (1994)
M.M. Burcin, G. Schiedner, S. Kochanek, S.Y. Tsai, B.W. O’Malley, Proc. Natl Acad. Sci. USA 96, 355–360 (1999)
R.V. Abruzzese, D. Godin, V. Mehta, J.L. Perrard, M. French, W. Nelson, G. Howell, M. Coleman, B.W. O’Malley, J.L. Nordstrom, Mol. Ther. 2, 276–287 (2000)
J.L. Nordstrom, Steroids 68, 1085–1094 (2003)
T. Kafri, H. van Praag, F.H. Gage, I.M. Verma, Mol. Ther. 1, 516–521 (2000)
O. Sirin, F. Park, Gene 323, 67–77 (2003)
B. Mitta, C.C. Weber, M. Rimann, M. Fussenegger, Nucleic Acids Res. 32, e106 (2004)
S.C. Beutelspacher, N. Ardjomand, P.H. Tan, G.S. Patton, D.F. Larkin, A.J. George, M.O. McClure, Exp. Eye Res. 80, 787–794 (2005)
F. Galimi, E. Saez, J. Gall, N. Hoong, G. Cho, R.M. Evans, I.M. Verma, Mol. Ther. 11, 142–148 (2005)
S. Hartenbach, M. Fussenegger, J. Biotechnol. 120, 83–98 (2005)
H.L. Heine, H.S. Leong, F.M. Rossi, B.M. McManus, T.J. Podor, Methods Mol. Med. 112, 109–154 (2005)
B. Mitta, C.C. Weber, M. Fussenegger, J. Gene Med. 7, 1400–1408 (2005)
K. Okamoto, J. Fujisawa, M. Reth, S. Yonehara, Genes Cells 11, 177–191 (2006)
W. Weber, W. Bacchus, F. Gruber, M. Hamberger, M. Fussenegger, J. Biotechnol. 131, 150–158 (2007)
H. Hurttila, J.K. Koponen, E. Kansanen, H.K. Jyrkkanen, A. Kivela, R. Kylatie, S. Yla-Herttuala, A.L. Levonen, Gene Ther. 15, 1271–1279 (2008)
S. Goverdhana, M. Puntel, W. Xiong, J.M. Zirger, C. Barcia, J.F. Curtin, E.B. Soffer, S. Mondkar, G.D. King, J. Hu, S.A. Sciascia, M. Candolfi, D.S. Greengold, P.R. Lowenstein, M.G. Castro, Mol. Ther. 12, 189–211 (2005)
Y. Kawano, T. Yoshida, K. Hieda, J. Aoki, H. Miyoshi, Y. Koyanagi, J. Virol. 78, 11352–11359 (2004)
H. Kuwata, Y. Watanabe, H. Miyoshi, M. Yamamoto, T. Kaisho, K. Takeda, S. Akira, Blood 102, 4123–4129 (2003)
H. Ebina, J. Aoki, S. Hatta, T. Yoshida, Y. Koyanagi, Microbes Infect. 6, 715–724 (2004)
U.K. von Schwedler, M. Stuchell, B. Muller, D.M. Ward, H.Y. Chung, E. Morita, H.E. Wang, T. Davis, G.P. He, D.M. Cimbora, A. Scott, H.G. Krausslich, J. Kaplan, S.G. Morham, W.I. Sundquist, Cell 114, 701–713 (2003)
H. Miyoshi, U. Blomer, M. Takahashi, F.H. Gage, I.M. Verma, J. Virol. 72, 8150–8157 (1998)
A. Adachi, H.E. Gendelman, S. Koenig, T. Folks, R. Willey, A. Rabson, M.A. Martin, J. Virol. 59, 284–291 (1986)
Z.L. Xu, H. Mizuguchi, T. Mayumi, T. Hayakawa, Gene 309, 145–151 (2003)
H.R. Jayakar, M.A. Whitt, J. Virol. 76, 8011–8018 (2002)
M.R. Mautino, R.A. Morgan, Aids Patient Care STDS 16, 11–26 (2002)
M. Mukhtar, H. Duke, M. BouHamdan, R.J. Pomerantz, Hum. Gene Ther. 11, 347–359 (2000)
M.R. Mautino, R.A. Morgan, Gene Ther. 9, 421–431 (2002)
A. Banerjea, M.J. Li, G. Bauer, L. Remling, N.S. Lee, J. Rossi, R. Akkina, Mol. Ther. 8, 62–71 (2003)
M.J. Li, G. Bauer, A. Michienzi, J.K. Yee, N.S. Lee, J. Kim, S. Li, D. Castanotto, J. Zaia, J.J. Rossi, Mol. Ther. 8, 196–206 (2003)
H. Nishitsuji, T. Ikeda, H. Miyoshi, T. Ohashi, M. Kannagi, T. Masuda, Microbes Infect. 6, 76–85 (2004)
R. Zufferey, J.E. Donello, D. Trono, T.J. Hope, J. Virol. 73, 2886–2892 (1999)
E. Vigna, S. Cavalieri, L. Ailles, M. Geuna, R. Loew, H. Bujard, L. Naldini, Mol. Ther. 5, 252–261 (2002)
R. Vogel, L. Amar, A.D. Thi, P. Saillour, J. Mallet, Hum. Gene Ther. 15, 157–165 (2004)
Acknowledgments
We thank Hiroyuki Miyoshi (RIKEN BioResource Center) for providing pCMV-VSV-G-RSV-Rev and pCAG-HIVgp, Elisa Izaurralde (European Molecular Biology Laboratory) for EGFP-fused VSV M-expressing plasmid, and Wesley Sundquist (Department of Biochemistry, University of Utah) for dominant-negative mutant VPS4B-expressing plasmid. We are also grateful to Joanne Martin for proofreading of the manuscript and members of the Laboratory of Viral Pathogenesis and the Laboratory for Host Factors for support of experimental techniques and helpful discussions. This work was supported by grants from the Ministry of Health, Labour and Welfare and the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Shinoda, Y., Hieda, K., Koyanagi, Y. et al. Efficient transduction of cytotoxic and anti-HIV-1 genes by a gene-regulatable lentiviral vector. Virus Genes 39, 165–175 (2009). https://doi.org/10.1007/s11262-009-0382-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-009-0382-x