
Abstract
The complete DNA sequence of the mildly virulent Gallid herpesvirus type 2 strain CU-2 was determined and consists of 176,922 bp with an overall gene organization typical of class E herpesviruses. Phylogenetically, this strain partitions in its own branch between the virulent strains RB-1B, Md11, and Md5, and the vaccine strain CVI988. Overall, the genome of CU-2 is more similar to that of CVI988, with identically sized unique short regions of 11,651 bp. As in CVI988, an insertion of 177 bp was identified in the overlapping genes encoding the Meq, RLORF6, and 23 kDa proteins within the repeat long region of the genome. A total of 15 single nucleotide polymorphisms (SNPs) common to both CU-2 and CVI988, and not occurring in virulent strains, were identified in the genes encoding UL29, UL45, UL50, UL52, LORF10, RLORF14a, RLORF12, Meq(RLORF7), 23kDa, ICP4, US3, and two hypothetical proteins MDV071.4 and MDV076.4. Each gene encoding UL29 and Meq contained two SNPs. Only one major open reading frame (ORF) encoding UL41, the virus host shutoff (VHS) ribonuclease, was disrupted in the CU-2 genome. An additional cytosine after the 25 codon is predicted to produce a truncated protein of 97 aa. Since GaHV-2 mutants lacking UL41 have been reported to retain their virulence, other factors are likely responsible for the low virulence of CU-2. It is largely suspected that SNPs in common with CVI988 along with the insertions in the Meq loci are responsible for its phenotype. Conversely, we identified 43 nonsynonymous mutations (within 23 genes) that may contribute to the virulence of CU-2. These SNPs are shared exclusively with all sequenced virulent strains (Md5, Md11, and RB-1B) and not present within the CVI988 genome. Although most occur in proteins of unknown function, a significant percentage is in proteins involved in virion assembly.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Gallid herpesvirus type 2 is an alphaherpesvirus that causes a lymphoproliferative disease (Marek’s disease, MD) in its natural host, the chicken. GaHV-2 (also known as MDV-1) belongs to the genus Mardivirus, which also contains two closely related but distinct species: GaHV-3, a nononcogenic herpesvirus also known as MDV-2, and turkey herpesvirus 1 (HVT) or Meleagrid herpesvirus type 1 (MeHV-1), previously MDV-3. Selected GaHV-2 and MeHV-1 strains, including attenuated variants of GaHV-2, have been used effectively as vaccines to protect chickens against tumor induction since the late 1960s [6, 30, 34, 40]. Despite the efforts to control MD through mass vaccination strategies, there is evidence that GaHV-2 has evolved to greater virulence during the last 40 years [55]. Whether this was the result of mass vaccination or independent of it remains a topic of debate. Nevertheless there is great concern within the poultry industry that current vaccines will fail to protect against these evolving virulent field isolates. Although both clinical and laboratory data agree that field isolates have evolved to greater virulence, the molecular basis for this evolution is still unknown. A classification nomenclature based on tumor response in vaccinated and control chickens has proposed five GaHV-2 pathotypes: attenuated (a), mildly virulent (m), virulent (v), very virulent (vv), and very virulent plus (vv+) [54, 56]. Unfortunately, little progress has been made correlating pathotype (or phenotype) with genotype. This is a formidable task since the genome of GaHV-2 is 180 kb [25, 26, 53].
Like other alphaherpesviruses, the GaHV-2 genome is a linear double-stranded molecule, which is organized (class E genome) identically to that of the prototype of the virus subfamily, herpes simplex virus type 1 (HSV-1) [36]. Class E genomes consist of two unique sequences (unique long, UL and unique short, US) each of which is flanked by inverted repeat sequences (TRL/IRL and TRS/IRS), respectively [35]. During DNA replication, the UL and US regions can flip-flop relative to each other, and consequently four forms of the viral genomes exist [35, 37]. In cell cultures (chick or duck embryo fibroblast), the genome is linear (packaged form), circular, or concatameric (replicative form). In lymphoblastoid cell lines established from MD tumors, the viral genomes are maintained as circular episomes [16, 33, 52] or integrated into host chromosomes [9, 10, 21]. Sequencing of GaHV-2 genomes using Sanger dideoxy chain termination has identified >140 open reading frames (ORFs) [31], the majority of which are present in unique regions and share substantial homology with collinear genes within the genomes of other alphaherpesviruses. Unique mardiviral ORFs are predominately found in the repeat regions and immediate adjacent regions (junctions) extending into the unique regions. These junctions have been a “hotspot” for integration of retro- and poxviral sequences [1, 2, 8, 13, 17–19, 23]. To date, the complete DNA sequences of five strains of GaHV-2 are present in GenBank [26, 29, 49, 50, 53], four of which are DNA sequences from viruses classified as virulent (v) or very virulent (vv). We have previously reported the sequence of the attenuated vaccine strain CVI988 (Rispens) of GaHV-2, the most commonly used vaccine strain worldwide. We now present the nucleotide sequence of a mildly virulent strain known as CU-2. This strain was originally isolated and cloned by Smith and Calnek [45] and pathotyped as mildly virulent (mv) in susceptible chickens [3, 14, 46]. To identify mutations, which may be responsible for its less virulent phenotype, CU-2 ORFS were compared with those found within the genomes of the attenuated strain CVI988 [34, 49] and the very virulent strains Md5, Md11, and RB-1B [26, 50, 53]. The sequencing of the CU-2 strain was done using pyrosequencing offered by 454 Life Sciences Corp. with substantial savings in costs and time.
Materials and methods
Viruses, cells, and media
The CU-2 strain (passage 14) of GaHV-2 was obtained from K. Schat (Cornell University, Ithaca, NY). Chick embryo fibroblasts (CEFs) were isolated from 9-day-old embryos, and maintained in minimal essential medium (MEM) supplemented with 8.0% fetal bovine serum (FBS) and antibiotics. CU-2 was propagated on CEFs in MEM supplemented with 2.0% FBS and antibiotics.
Purification of DNA from CU-2-infected cells
CU-2-infected CEFs were harvested at 5 days postinfection when the cytopathic effect was close to 80%. Cells were trypsinized and pelleted at 300 × g at 4°C for 10 min. The supernatant was removed and the cells were resuspended in 20 ml of wash buffer (150 mM sodium chloride, 10 mM Tris pH 8.0, and 5 mM EDTA) and centrifuged at 300 × g for 5 min. The pellet was resuspended in hypotonic lysis solution (10 mM potassium chloride, 10 mM Tris pH 8.0, 5 mM EDTA, and 1.0% Triton X-100 and 36 mM β-mercaptoethanol). The suspension was placed on ice for 10 min. RNaseA was added to a final concentration of 50 μg/ml and the solution was incubated at 37°C for 30 min. Nucleocapsids were released from cellular debris by centrifugation at 1800 × g for 10 min. Nucleocapsids in the supernatant were further purified through a 30% sucrose cushion at 13,000 rpm at 4°C for 1 h using a SW28 Beckman ultracentrifuge rotor. Pelleted nucleocapsids were resuspended in 10 mM Tris pH 8.0, 3 mM EDTA, and proteinase K (2 mg/ml) and incubated at 37°C for 18 h. Proteins were extracted from viral DNA using phenol–chloroform extractions. The DNA was concentrated using ammonium acetate/ethanol precipitation and quantified at OD260. The quality of the viral DNA was assessed via BamHI restriction endonuclease digestion.
Determination of CU-2 genomic termini, 132 bp repeat, and a-like sequences
The terminal sequences were characterized from linear nucleocapsid DNA by ligation-mediated PCR. Briefly, CU-2 DNA was blunt-ended using Klenow DNA polymerase. A directional adapter of approximately 40 bp was ligated to the blunt-ended genomic DNA in high molar excess using T4 DNA ligase and the nicked-ligated DNA was repaired using Klenow DNA polymerase. The resulting DNA was subjected to PCR amplification with Platinum Taq HiFi (Invitrogen) using one primer specific to the adapter and one primer internal by several hundred base pairs to the predicted cleavage site on the 5′ end of the genome. The amplification was repeated, substituting a primer internal by several hundred base pairs to the predicted 3′ terminus. The PCR products were column purified from an agarose gel and cloned into the pCR2.1-TOPO vector (Invitrogen). The resulting clones were screened using colony PCR and sequenced using ABI-Sanger methodology. The methodology for determining both the copy numbers of the 132 bp repeats present in the repeat long regions of the CU-2 genome and the a-like sequences at the IRL/IRS junction was described previously [47, 48].
DNA sequencing
Sequencing of 5.0 μg of CU-2 DNA was carried out commercially using a pyrosequencing platform, the Genome Sequencer 20 (GS20) System (454 Life Science Corporation). This involved the construction of a random library of the CU-2 DNA using the methodology described by Margulies et al. [28]. Briefly, CU-2 DNA was sheared by nebulization to 300–700 bp. DNA ends were repaired and phosphorylated using T4 DNA polymerase and T4 polynucleotide kinase, respectively. Adaptor oligonucleotides were added to the repaired ends using T4 DNA ligase. Purified DNA containing adaptors was hybridized to DNA capture beads to ensure only one DNA fragment per bead and clonally amplified using emulsion PCR. After disruption of the emulsion bubbles, DNA capture beads were sieved onto a 40 × 75 mm PicoTiterPlate equipped with an eight-lane gasket and flooded with reagents needed for pyrosequencing. Sequence reads, contigs, and quality scores for sequences and contigs were obtained from 454 Life Sciences. Problematic regions containing homopolymers or repetitive regions (i.e., 132 bp repeats within the repeat long regions and a-like sequences) were sequenced from PCR products generated in reactions containing Platinum Taq DNA polymerase (Invitrogen, Carlsbad, CA) and numerous custom primers. PCR conditions were optimized using an Eppendorf Mastercycler Gradient to determine the optimal annealing temperatures and 5% DMSO was included in the reaction mixtures to increase product yields. ABI-Sanger based DNA sequencing of PCR products and clones was performed at the South Atlantic Area sequencing facility (Athens, GA) and Polymorphic DNA Technologies, Inc. (Alameda, CA) using the BigDye terminator cycle sequencing protocol and analyzed on a model ABI-3730 XL DNA Analyzer (Applied Biosystems, Foster City, CA).
DNA sequence analysis
CU-2 DNA sequences were assembled from 23,921 reads (average length 150 nuc.) using the Sequencher Program (Gene Codes, Ann Arbor, MI). The final sequence represents a 13-fold coverage at each base pair. Ambiguities in the CU-2 sequencing data were resolved by re-sequencing using ABI-Sanger methodology. DNA sequences were maintained using Lasergene (DNASTAR, Madison, WI) and analyzed using NCBI Entrez and other web-based tools. Homology searches were conducted using the NCBI programs blastP and PsiBlast [39] with default settings. Multiple alignments of proteins and nucleotide sequences were generated using MAFFT 6.5, MUSCLE, MultAlin, and MEGA 3.1 [7, 12, 22, 24]. Phylogenetic trees were drawn using Phylip [32]. The 132 bp repeat elements were investigated using the Tandyman program (Los Alamos National Laboratory, NM). The sequences of Md5, Md11, GA, CVI988, and RB-1B used for comparative genomic studies were obtained from GenBank [2, 26, 29, 50, 53]. The complete nucleotide sequence of CU-2 has been submitted to GenBank.
Results and discussion
454 Life Science pyrosequencing
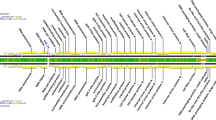
Using 454 sequencing technology, we determined the complete DNA sequence of the mildly virulent GaHV-2 strain known as CU-2. More than 99.0% coverage was obtained by assembling the raw sequence data to an overall average coverage of 13X. This low coverage for pyrosequencing was due to the high concentration of contaminating cellular DNA (62.72%) within the viral DNA prep (Fig. 1). Only 37.3% of the total run (64,173 reads) was CU-2 specific. Individual reads were initially assembled into large contigs representing the unique long (UL) and unique short (US) and repeat long (RL) and repeat short (RS) genomic segments. The final sequence contains identical inverted repeat regions (terminal repeat long; internal repeat long) and (internal repeat short; terminal repeat short) due to the lack of single nucleotide polymorphisms (SNPs) that could differentiate the paired inverted repeats. One of the shortcomings of pyrosequencing is its inability to resolve mononucleotide reiteration (i.e., homopolymers). Twenty-four homopolymer stretches (Fig. 2) had to be resolved using ABI-Sanger-based sequencing of PCR products. More than 80% of the homopolymer stretches were in A/T runs rather the C/G runs.

Visualization of an ethidium bromide-stained agarose gel containing BamHI-digested GaHV-2 DNA. Lane 1: molecular weight marker—the 2-log DNA ladder (New England Biolabs), lane 2: GaHV-2 strain 584A(BAC) and lane 3 GaHV-2 strain CU-2

Genomic location of homopolymers not resolved using 454 Life Sciences technology
Genome organization and phylogenetic relatedness
The complete CU-2 genome has a length of 176,922 bp and organized into 6 regions characteristic of class E herpesvirus. The UL region is 113,500 bp in length and extends from positions 13,289 to 126,788. The US region (pos 152,619–164,269) is 11,651 bp in length, a size identical to that found within the genome of the attenuated vaccine strain CVI988. The terminal repeat long (TRL) and internal repeat long (IRL) extends from positions 335 to 13,288 and 126,789 to 139,742 respectively and both are 12,954 bp in length. Both the internal repeat short (IRS) and terminal repeat short (TRS) are 12,299 bp in length and extend from positions 140,320 to 152,618 and 164,270 to 176,568, respectively. The a-like sequence, 577 bp in length bracketed by DR1 sequences (ggccgcgagagg), starts at position 139,743 and ends at position 140,319. The termini sequences are defined by the DR1 sequence (cctctcgcggcc) ending at position 334 and starting at position 176,569. CU-2 is the first GaHV-2 genome whose termini have been determined. The sequences of strains GA and Md11 are missing in the terminal a-like sequences and terminal repeat short sequence, respectively [26, 29]. Also, the sequences of CVI988 and RB-1B lack the terminal sequences since these genomes were sequenced from BAC (bacterial artificial chromosome) clones [49, 50].
As shown in Fig. 3 intra-strain sequence heterogeneity was observed at both termini in the CU-2 genome. The degree of the heterogeneity was unexpected and may be the result of imprecise cleavage as has been previously reported for the cleavage of the HHV-6 genome [11]. It is also possible that degradation is occurring at the ends of the viral DNA subsequent to cleavage. Whether this is happening during infection or as a byproduct of DNA isolation procedure is not clear. Because of these uncertainties the sequence of the CU-2 genome submitted to GenBank contains the longest terminal sequences (TermA-08a and Term B-06; Fig. 3).

Sequence length polymorphisms at the 5′ and 3′ termini of the CU-2 genome
In order to determine the evolutionary relatedness among GaHV-2 strains, individual sequences containing the UL, IRL, IRS, and US regions (approximately 152 Kb) were aligned using the web-based alignment program MAFFT [22]. Phylogenetically, CU-2 segregates between Md5 and CVI988 (Fig. 4). This was an expected result since pathotypically CU-2 is not fully attenuated and contains virulence factors in common with Md5, Md11, RB-1B, and the distantly related strain GA.

Phylogenetic relatedness of CU-2 with an attenuated strain (CVI988) and virulent strains (Md5, Md11, RB-1B, and GA)
Two regions that differ greatly among GaHV-2 strains are the TRL and IRL. These differences can be both intergenomic and intragenomic [38, 43, 47]. A 132-bp repeat motif is largely responsible for the variations. These motifs can vary from 2 copies per region to greater than 50 [43]. The expansion of the TRL and IRL regions occurs during serial passage of the virus in vitro and was thought to be somehow involved in attenuation. Generally, less virulent strains have a higher number of 132 bp repeats than more virulent ones. Within the repeat long regions of CU-2, only two copies of the 132 bp repeat are present. Interestingly, this is the same number of repeats that are found in early passage isolates of the avirulent vaccine strain CVI988. The role the 132 bp repeats play in virulence is puzzling [44]. At most they indicate the degree of passage or “passage history” that a strain has undergone in cell culture. It seems likely that other genetic changes besides the 132 bp expansion are responsible for attenuation [42].
Analysis of ORFs
ORFs within the CU-2 genome were annotated according to those found in the GaHV-2 genomes of strains GA, Md5, Md11, and RB-1B. A total of 197 CU-2 ORFs were compared with homologous ORFs in the genomes of these previously sequenced strains and an abbreviated list (72 ORFs) is presented in Table 1. ORFs that have 100% identity among all known sequenced GaHV-2 strains were omitted. Table 1 lists ORFs that differ among attenuated (CVI988) and virulent (Md5, Md11, and RB-1B) strains. Ninety-four percent of the differences are attributed to SNPs. Gross genetic alterations were identified in only seven genes: MDV054 and three diploid genes MDV004/MDV077, MDV005/MDV076, and MDV005.1/MDV077.5 encoding UL41, 23 kDa, RLORF7 (Meq), and RLORF6 proteins, respectively.
Relative to the UL41 ORF of other Mardiviruses, the CU-2 UL41 ORF contains an additional C residue within a stretch of C homopolymers 54 nucleotides downstream from the putative start codon (atcatgg). A truncated protein of 97 aa (MDV054a) is predicted due to the resulting frameshift. However, there are six start codons within 120 bp of the putative ATG. Two start codons are in frame with the MDV054a ORF. The use of three other potential start codons would generate polypeptides no larger than 24 aa. A polypeptide MDV054b would be generated using the remaining start codon (gatatga). This polypeptide (413 aa) would contain residues in common with the COOH terminus of UL41 and lack only 28 aa present at the amino terminus of full length UL41. This polypeptide would contain domains (XPG G-/I-region and BSM1) conserved among functional UL41 endoribonucleases [15, 20, 51]. Whether CU-2 expresses a functional UL41 is unknown. Since UL41 homologues are expressed from the genomes of related alphaherpesviruses and involved in viral host shutoff (VHS), a GaHV-2 strain containing a UL41 mutation might be attenuated. However, this is not the case. UL41 deletion mutants made in the Md5 (virulent) strain of GaHV-2 are just as virulent as wild-type virus (R.F. Silva, personal communication). Therefore it appears that UL41 of GaHV-2 is unlikely to be involved in virulence.
Three other genes encoding RLORF7 (or Meq), 23 kDa and RLORF6 within the “Meq loci” are far more likely to be involved in virulence. These genes overlap one another and are present in both repeat long regions (diploid). In the CU-2 genome these ORFs contain insertional polymorphisms identical to those found in some clones of the attenuated vaccine strain CVI988 [4, 27, 41, 47]. The insertions in RLORF7 are postulated to be the result of a proline-rich domain duplication [41]. Interestingly, proteins containing these duplications have been reported to exhibit a higher level of transrepression than Meq proteins of virulent pathotypes lacking the duplication [5]. Therefore it seems likely that the Meq loci contribute to the low virulence phenotype of CU-2.
Single nucleotide polymorphisms
Sequence analysis of CU-2 indicates that its genome is very similar to that of the attenuated strain CVI988. However, biologically, CU-2 is more virulent than CVI988 in susceptible chickens. The question remains what other factors may contribute to the mildly virulent phenotype? Are there SNPs that make CU-2 more virulent than CVI988? To address this question, we have identified polymorphisms in CU-2 ORFs that are common among virulent isolates and are different from attenuated strain CVI988. Table 2 contains a list of SNPs unique to CVI988 and absent from CU-2 or virulent strains. A total of 43 nonsynonymous mutations within 23 genes are listed. Although most occur in hypothetical proteins, a large percentage occurs in proteins involved in virion assembly. These include ORFs 31, 33, 38, 50, 51, 59, 60, 62, and 67 encoding homologues of herpes simplex virus type 1 UL19, UL21, UL26, UL37 UL38, UL46, UL47 UL49, and UL53, respectively. Two genes, MDV060 and MDV062 encoding tegument proteins UL47 (VP13/14) and UL49 (VP22), respectively, contain the greatest number of mutations. Most substitutions occur in residues that are not functionally equivalent and probably affect the protein’s function. Therefore, proteins containing the mutations listed in Table 2 are suspected to play a potential role in virulence. The last question that was addressed in the genomic comparison between CU-2 and Md5, Md11, RB-1B, and CVI988 relates to SNPs shared between CU2 and CVI988 and not occurring in virulent strains. Collectively these are referred to as “avirulent SNPs.” A total of 15 SNPs in 13 ORFs were identified (Table 3). All but two (MDV071.4 and MDV076.4) occur in ORFs encoding proteins of known function. More than half of the SNPs occur in ORFs largely unique to GaHV-2 and not found in other alphaherpesviruses. These include ORFs 57.8, 71, 71.4, 73, 74, 76, and 76.4. The most noteworthy avirulent SNPs are the Ser74 and Glu77 with MDV076 encoding the oncoprotein Meq. These same mutations have been identified in other less virulent strains of GaHV-2 [41, 47].
Conclusion
454 Life Sciences Corp. pyrosequencing allowed the rapid and cost-effective sequencing of a Gallid herpesvirus type 2 strain (CU-2). The complete sequence with 13-fold coverage was estimated to cost 1.4 U.S. cents per base pair. This is a substantial savings compared to Sanger-based methods which are estimated to cost 3.9–5.6 U.S. cents per base pair. Moreover, this technique does not require the laborious step of generating cloned DNA libraries and their associated plasmid purification costs. However, pyrosequencing does have a minor drawback in its inability to accurately determine the number of nucleotides in homopolymer stretches. In order to resolve these regions (Fig. 2), PCR products were generated and subjected to ABI-Sanger sequencing technology. The genomic comparison between the mildly attenuated strain CU-2 and other published sequences of GaHV-2 strains has identified both gross and subtle genetic changes. CU-2 contains an insertion of 177 bp within the loci present in the repeat long regions of the genome encoding the three overlapping genes MDV004/MDV077, MDV005.1/MDV077.5 and MDV005/76 encoding the 23 kDa and RLORF6 proteins, and Meq oncoproteins, respectively. These are the same insertional mutations found in the attenuated vaccine strain CVI988 and other lower virulent strains. Besides a mutation causing a frameshift in the UL41 gene (encoding VHS), most mutations in CU-2 ORFs were nonsynonymous amino acid substitutions. These substitutions were divided into groups based on whether they were shared among ORFs within the genomes of virulent strains or those in attenuated strains. The identification of these mutations will further our understanding of genes involved in virulence and allow for the development of pathotyping tests based on sequencing data in place of time-consuming bioassays which use hundreds of birds.
References
R. Borenshtein, I. Davidson, Development of the hot spot-combined PCR assay for detection of retroviral insertions into Marek’s disease virus. J. Virol. Methods 82, 119–127 (1999)
P. Brunovskis, L.F. Velicer, The Marek’s disease virus (MDV) unique short region: alphaherpesvirus-homologous, fowlpox virus-homologous, and MDV-specific genes. Virology 206, 324–338 (1995)
B.W. Calnek, J. Fabricant, K.A. Schat, K.K. Murthy, Pathogenicity of low-virulence Marek’s disease viruses in normal versus immunologically compromised chickens. Avian Dis. 21, 346–358 (1977)
K.S. Chang, K. Ohashi, M. Onuma, Diversity (polymorphism) of the Meq gene in the attenuated Marek’s disease virus (MDV) serotype 1 and MDV-transformed cell lines. J. Vet. Med. Sci. 64, 1097–1101 (2002)
K.S. Chang, K. Ohashi, M. Onuma, Suppression of transcription activity of the MEQ protein of oncogenic Marek’s disease virus serotype 1 (MDV1) by L-MEQ of non-oncogenic MDV1. J. Vet. Med. Sci. 64, 1091–1095 (2002)
A.E. Churchill, L.N. Payne, R.C. Chubb, Marek’s disease immunization against Marek’s disease using a live attenuated virus. Lancet 1, 610–611 (1969)
F. Corpet, Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 16, 10881–10890 (1988)
I. Davidson, R. Borenshtain, In vivo events of retroviral long terminal repeat integration into Marek’s disease virus in commercial poultry: detection of chimeric molecules as a marker. Avian Dis. 45, 102–121 (2001)
H.J. Delecluse, W. Hammerschmidt, Status of Marek’s disease virus in established lymphoma cell lines: herpesvirus integration is common. J. Virol. 67, 82–92 (1993)
H.J. Delecluse, S. Schuller, W. Hammerschmidt, Latent Marek’s disease virus can be activated from its chromosomally integrated state in herpesvirus-transformed lymphoma cells. EMBO J. 12, 3277–3286 (1993)
H. Deng, S. Dewhurst, Functional identification and analysis of cis-acting sequences which mediate genome cleavage and packaging in human herpesvirus 6. J. Virol. 72, 320–329 (1998)
R.C. Edgar, MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004)
D. Endoh, M. Ito, K.O. Cho, Y. Kon, T. Morimura, M. Hayashi, M. Kuwabara, Retroviral sequence located in border region of short unique region and short terminal repeat of Md5 strain of Marek’s disease virus type 1. J. Vet. Med. Sci. 60, 227–235 (1998)
J. Fabricant, M. Ianconescu, B.W. Calnek, Comparative effects of host and viral factors on early pathogenesis of Marek’s disease. Infect. Immun. 16, 136–144 (1977)
M. Hohl, I. Dunand-Sauthier, L. Staresincic, P. Jaquier-Gubler, F. Thorel, M. Modesti, S.G. Clarkson, O.D. Scharer, Domain swapping between FEN-1 and XPG defines regions in XPG that mediate nucleotide excision repair activity and substrate specificity. Nucleic Acids Res. 35, 3053–3063 (2007)
R.J. Isfort, D. Robinson, H.J. Kung, Purification of genomic sized herpesvirus DNA using pulse-field electrophoresis. J. Virol. Methods 27, 311–317 (1990)
R.J. Isfort, Z. Qian, D. Jones, R.F. Silva, R. Witter, H.J. Kung, Integration of multiple chicken retroviruses into multiple chicken herpesviruses: herpesviral gD as a common target of integration. Virology 203, 125–133 (1994)
D. Jones, R. Isfort, R. Witter, R. Kost, H.J. Kung, Retroviral insertions into a herpesvirus are clustered at the junctions of the short repeat and short unique sequences. Proc. Natl. Acad. Sci. USA 90, 3855–3859 (1993)
D. Jones, P. Brunovskis, R. Witter, H.J. Kung, Retroviral insertional activation in a herpesvirus: transcriptional activation of US genes by an integrated long terminal repeat in a Marek’s disease virus clone. J. Virol. 70, 2460–2467 (1996)
K. Karbstein, J.A. Doudna, GTP-dependent formation of a ribonucleoprotein subcomplex required for ribosome biogenesis. J. Mol. Biol. 356, 432–443 (2006)
C. Kaschka-Dierich, K. Nazerian, R. Thomssen, Intracellular state of Marek’s disease virus DNA in two tumour-derived chicken cell lines. J. Gen. Virol. 44, 271–280 (1979)
K. Katoh, K. Kuma, H. Toh, T. Miyata, MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33, 511–518 (2005)
R. Kost, D. Jones, R. Isfort, R. Witter, H.J. Kung, Retrovirus insertion into herpesvirus: characterization of a Marek’s disease virus harboring a solo LTR. Virology 192, 161–169 (1993)
S. Kumar, K. Tamura, M. Nei, MEGA3: integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief Bioinform. 5, 150–163 (2004)
L.F. Lee, E.D. Kieff, S.L. Bachenheimer, B. Roizman, P.G. Spear, B.R. Burmester, K. Nazerian, Size and composition of Marek’s disease virus deoxyribonucleic acid. J. Virol. 7, 289–294 (1971)
L.F. Lee, P. Wu, D. Sui, D. Ren, J. Kamil, H.J. Kung, R.L. Witter, The complete unique long sequence and the overall genomic organization of the GA strain of Marek’s disease virus. Proc. Natl. Acad. Sci. USA 97, 6091–6096 (2000)
S.I. Lee, M. Takagi, K. Ohashi, C. Sugimoto, M. Onuma, Difference in the Meq gene between oncogenic and attenuated strains of Marek’s disease virus serotype 1. J. Vet. Med. Sci. 62, 287–292 (2000)
M. Margulies, M. Egholm, W.E. Altman, S. Attiya, J.S. Bader, L.A. Bemben, J. Berka, M.S. Braverman, Y.J. Chen, Z. Chen, S.B. Dewell, L. Du, J.M. Fierro, X.V. Gomes, B.C. Godwin, W. He, S. Helgesen, C.H. Ho, G.P. Irzyk, S.C. Jando, M.L. Alenquer, T.P. Jarvie, K.B. Jirage, J.B. Kim, J.R. Knight, J.R. Lanza, J.H. Leamon, S.M. Lefkowitz, M. Lei, J. Li, K.L. Lohman, H. Lu, V.B. Makhijani, K.E. McDade, M.P. McKenna, E.W. Myers, E. Nickerson, J.R. Nobile, R. Plant, B.P. Puc, M.T. Ronan, G.T. Roth, G.J. Sarkis, J.F. Simons, J.W. Simpson, M. Srinivasan, K.R. Tartaro, A. Tomasz, K.A. Vogt, G.A. Volkmer, S.H. Wang, Y. Wang, M.P. Weiner, P. Yu, R.F. Begley, J.M. Rothberg, Genome sequencing in microfabricated high-density picolitre reactors. Nature 437, 376–380 (2005)
M. Niikura, J. Dodgson, H. Cheng, Direct evidence of host genome acquisition by the alphaherpesvirus Marek’s disease virus. Arch. Virol. 151, 537–549 (2005)
W. Okazaki, H.G. Purchase, B.R. Burmester, Protection against Marek’s disease by vaccination with a herpesvirus of turkeys. Avian Dis. 14, 413–429 (1970)
K. Osterrieder, J.-F. Vautherot, The genome content of Marek’s disease-like viruses, in Marek’s Disease: An Emerging Problem, ed. by F. Davison, V. Nair (Elsevier, Oxford, UK, 2004)
J.D. Retief, Phylogenetic analysis using PHYLIP. Methods Mol. Biol. 132, 243–258 (2000)
H.-J. Rhiza, B. Bauer, Circular forms of viral DNA in Marek’s disease virus-transformed lymphoblastoid cells. Arch. Virol. 72, 211–216 (1982)
B.H. Rispens, H. van Vloten, N. Mastenbroek, H.J. Maas, K.A. Schat, Control of Marek’s disease in the Netherlands. I. Isolation of an avirulent Marek’s disease virus (strain CVI 988) and its use in laboratory vaccination trials. Avian Dis. 16, 108–125 (1972)
B. Roizman, in The Herpesviruses, ed. by B. Roizman (Plenum, London, 1982), p. 1–23
B. Roizman, in Fields Virology, 4th edn., ed. by B.N. Fields, D.M. Knipe, P.M. Howley (Lippincott-Raven Press, New York, 1996), p. 2221–2230
B. Roizman, A.E. Sears, in Fields Virology, ed. by B.N. Fields, D.M. Knipe, P.M. Howley (Lippincott-Raven Press, New York, 1996), p. 2231–2295
N. Ross, M.M. Binns, M. Sanderson, K.A. Schat, Alterations in DNA sequence and RNA transcription of the BamHI-H fragment accompany attenuation of oncogenic Marek’s disease herpesvirus. Virus Genes 7, 33–51 (1993)
A.A. Schaffer, L. Aravind, T.L. Madden, S. Shavirin, J.L. Spouge, Y.I. Wolf, E.V. Koonin, S.F. Altschul, Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucleic Acids Res. 29, 2994–3005 (2001)
K.A. Schat, B.W. Calnek, Characterizations of an apparently non-oncogenic Marek’s disease virus. J. Natl. Cancer Inst. 60, 1141–1146 (1978)
C.E. Shamblin, N. Greene, V. Arumugaswami, R.L. Dienglewicz, M.S. Parcells, Comparative analysis of Marek’s disease virus (MDV) glycoprotein-, lytic antigen pp38- and transformation antigen Meq-encoding genes: association of Meq mutations with MDVs of high virulence. Vet. Microbiol. 102, 147–167 (2004)
R.F. Silva, I. Gimeno, Oncogenic Marek’s disease viruses lacking the 132 base pair repeats can still be attenuated by serial in vitro cell culture passages. Virus Genes 34, 87–90 (2007)
R.F. Silva, R.L. Witter, Genomic expansion of Marek’s disease virus DNA is associated with serial in vitro passage. J. Virol. 54, 690–696 (1985)
R.F. Silva, S.M. Reddy, B. Lupiani, Expansion of a unique region in the Marek’s disease virus genome occurs concomitantly with attenuation but is not sufficient to cause attenuation. J. Virol. 78, 733–740 (2004)
M.W. Smith, B.W. Calnek, Effect of virus pathogenicity on antibody production in Marek’s disease. Avian Dis. 17, 727–736 (1973)
M.W. Smith, B. Calnek, Comparative features of low-virulence and high-virulence Marek’s disease infections. Avian Pathol. 3, 229–246 (1974)
S.J. Spatz, R.F. Silva, Polymorphisms in the repeat long regions of oncogenic and attenuated pathotypes of Marek’s disease virus 1. Virus Genes 35, 41–53 (2007)
S.J. Spatz, R.F. Silva, Sequence determination of variable regions within the genomes of gallid herpesvirus-2 pathotypes. Arch. Virol. 152, 1665–1678 (2007)
S.J. Spatz, L. Petherbridge, Y. Zhao, V. Nair, Comparative full-length sequence analysis of oncogenic and vaccine (Rispens) strains of Marek’s disease virus. J. Gen. Virol. 88, 1080–1096 (2007)
S.J. Spatz, Y. Zhao, L. Petherbridge, L.P. Smith, S.J. Baigent, V. Nair, Comparative sequence analysis of a highly oncogenic but horizontal spread-defective clone of Marek’s disease virus. Virus Genes 35, 753–766 (2007)
B. Taddeo, B. Roizman, The virion host shutoff protein (UL41) of herpes simplex virus 1 is an endoribonuclease with a substrate specificity similar to that of RNase A. J. Virol. 80, 9341–9345 (2006)
A. Tanaka, S. Silver, M. Nonoyama, Biochemical evidence of the nonintegrated status of Marek’s disease virus DNA in virus-transformed lymphoblastoid cells of chicken. Virology 88, 19–24 (1978)
E.R. Tulman, C.L. Afonso, Z. Lu, L. Zsak, D.L. Rock, G.F. Kutish, The genome of a very virulent Marek’s disease virus. J. Virol. 74, 7980–7988 (2000)
R.L. Witter, Characteristics of Marek’s disease viruses isolated from vaccinated commercial chicken flocks: association of viral pathotype with lymphoma frequency. Avian Dis. 27, 113–132 (1983)
R.L. Witter, Increased virulence of Marek’s disease virus field isolates. Avian Dis. 41, 149–63 (1997)
R.L. Witter, B.W. Calnek, C. Buscaglia, I.M. Gimeno, K.A. Schat, Classification of Marek’s disease viruses according to pathotype: philosophy and methodology. Avian Pathol. 34, 75–90 (2005)
Acknowledgments
We thank Jeremy Volkening for his contributions in all stages of this research. Jeremy has outstanding skills in bioinformatics, scientific writing, and computer graphics. This research was funded by the United States Department of Agriculture CRIS program (project number 6612-32000-043).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Spatz, S.J., Rue, C.A. Sequence determination of a mildly virulent strain (CU-2) of Gallid herpesvirus type 2 using 454 pyrosequencing. Virus Genes 36, 479–489 (2008). https://doi.org/10.1007/s11262-008-0213-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-008-0213-5