
Abstract
Murray Valley encephalitis virus (MVEV) is a medically important mosquito-borne flavivirus found in Australia and Papua New Guinea (PNG). Partial envelope gene nucleotide sequences of 28 isolates of MVEV from Western Australia (WA) between 1972 and 2003 were aligned and compared phylogenetically with the prototype MVE-1-51 from Victoria in 1951 and isolates from northern Queensland and PNG. Monoclonal antibody-binding patterns were also investigated. Results showed that the majority of isolates of MVEV from widely disparate locations in WA were genetically and phenotypically homogeneous. Furthermore, isolates of MVEV from WA and northern Queensland were almost identical, confirming results from earlier studies. Recent isolates of MVEV from Western Province in PNG were more similar to Australian isolates of MVEV than to isolates from PNG in 1956 and 1966, providing further evidence for the movement of flaviviruses between PNG and Australia. Additional representatives of a unique variant of MVEV (OR156) from Kununurra in the northeast Kimberley region of WA were also detected. This suggests that the OR156 lineage is still intermittently active but may be restricted to a small geographic area in northern WA, possibly due to altered biological characteristics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Murray Valley encephalitis virus (family Flaviviridae, genus Flavivirus, MVEV) is a medically important flavivirus transmitted by mosquitoes in Australia and Papua New Guinea (PNG) [1]. MVEV primarily exists in a transmission cycle between Culex annulirostris and birds. Humans are generally considered to be dead-end hosts [2]. MVEV is enzootic in northern regions of Australia, and is occasionally responsible for widespread outbreaks of disease [1]. In recent years, MVEV activity has been detected as far south as the Midwest region of Western Australia (WA) [3, 4]. Approximately 1:1000 infections with MVEV in humans lead to disease [5], although the clinical to subclinical infection rate is often higher in epizootic areas [6, C. Johansen and A. Broom, unpublished results]. Disease ranges from mild febrile illness to potentially fatal encephalitis. Of those that develop encephalitis, 25% of sufferers will die, and 50% experience permanent neurological sequelae [2, 4].
A prospective surveillance programme for MVEV has been in place in WA since the mid-1970s that primarily involves regular bleeding of sentinel chicken flocks in northern WA. Serum is tested for the presence of antibodies to MVEV and the closely related Kunjin virus (KUNV) [7]. Results of the surveillance programme indicate that MVEV is enzootic in the Kimberley region [8]. However, MVEV occasionally moves further south into the Pilbara, Gascoyne, Murchison and Midwest regions when conditions become suitable [3].
In addition to surveillance using sentinel chicken flocks, mosquito collection trips are undertaken annually during the latter part of the wet season in the Kimberley region. Opportunistic mosquito collections are conducted in other regions south of the Kimberley, generally following the detection of seroconversions to MVEV in sentinel chickens. Mosquitoes are processed for virus isolation to determine infection rates in mosquitoes and to investigate which mosquito species are likely to be involved in MVEV transmission [9].
Murray Valley encephalitis virus isolates have been obtained from mosquitoes collected in northern WA since monitoring for arbovirus activity commenced in WA in 1972. The regular isolations of MVEV from mosquitoes in northern WA during the course of the surveillance programme facilitated a study to investigate genetic and phenotypic relationships and patterns of virus movement over a 32-year period.
Materials and methods
Nucleotide sequences obtained from part of the envelope (E) gene of representative isolates of MVEV (Table 1) from mosquitoes in the Kimberley, Pilbara and Gascoyne regions of northern WA were compared with other isolates of MVEV from Queensland [11] and Victoria [12] in Australia, and PNG [13, 14]. The E region was chosen because it has been used previously for phylogenetic analysis of flaviviruses [15–19]. The region used in this study corresponded to nucleotides 1454–1916 in the complete genome and E gene nucleotide 484 (amino acid 161) to nucleotide 945 (amino acid 315) of MVE-1-51 (AF161266) [20]. Major neutralisation regions in domains I and II of E [21], amino acid substitutions found in neutralising escape mutants [22] and other non-conservative amino acid substitutions previously identified in PNG isolates of MVEV from the 1960s [23] were located within the sequence region. Western Australian isolates included in the study represented each year and geographic location that MVEV was obtained from mosquitoes (Table 1, Fig. 1).

Location of origin of isolates of Murray Valley encephalitis virus used in the phylogenetic and phenotypic analyses
Total RNA was extracted from 140 μl of tissue culture supernatant or 10% suckling mouse brain suspensions of viral isolates of MVEV using a QIAamp Viral RNA kit (Qiagen Pty., Ltd., Clifton Hill, Victoria, Australia) or TRIZOL reagent (Gibco BRL Life Technologies, Gaithersburg, MD, USA) according to the manufacturer’s instructions and stored at −70°C. A one-step reverse transcription (RT) -polymerase chain reaction (PCR) was used to amplify a 675 bp region in the E gene of MVEV using MVE2F forward (5'-GGGGAGACTTATCTTACCTGAGA-3') and MVE2B reverse (5'-GGCGTCATGTCATTGAGACTTGC-3') primers (M. Poidinger, The University of Queensland). Between 1 and 5 μl of RNA was added to 0.2 ml thin-walled PCR tubes (Sarstedt, Nümbrecht, Germany) containing 100 ng of each primer, 200 μm dNTPs (Fisher Biotech, Subiaco, WA, Australia), 2.0 mM MgCl2 (Promega, Madison, WI, USA), 5.0 mM dithiothreitol (Promega), 5 units of avian myeloblastosis virus reverse transcriptase (Promega), 1.5 units of Taq polymerase (Promega), and 8 units of RNasin (Promega) in Taq buffer and Baxter water (Baxter Healthcare Pty., Ltd., Old Toongabbie, NSW, Australia) in a final reaction volume of 15 μl. Thermocycling (GeneAmp® PCR System 9700, Applied Biosystems Pty., Ltd., Scorseby, Victoria, Australia) conditions entailed one cycle of reverse transcription at 48°C for 30 min, followed by denaturation at 94°C for 2 min, then 30 cycles of 94°C for 1 min, annealing at 57°C for 30 s and elongation at 72°C for 1 min. The resulting RT-PCR products were purified after agarose gel electrophoresis (QIAquick Gel Extraction Kit, Qiagen). PCR products were directly sequenced in both directions using the BigDye system (Perkin Elmer Biosystems, Foster City, CA, USA) by the Australian Neuromuscular Research Institute Sequencing Service (Sir Charles Gairdner Hospital, Nedlands, WA, Australia) or the DNA Sequencing Services (Western Australian Genome Centre, Department of Clinical Immunology, Royal Perth Hospital, Perth, WA, Australia).
The E gene sequences were submitted to GenBank (EF015041 to EF015074) and compared with other available sequences of MVEV from Australia (MVE-1-51) and PNG (NG156 and NG6684) (EF015076 and EF015075). Multiple nucleotide sequence alignments were performed using the CLUSTAL W programme [24] using BioManager, provided by the Australian National Genome Information Service (http://www.angis.org.au). A 462 bp region of the alignment was used to complete phylogenetic analyses. MEGA 3.1 [25] was used to analyse the composition of the nucleotide sequence alignment. A hierarchical likelihood ratio test (hLRT) implemented in Modeltest 3.7 [26] in conjunction with PAUP* 4.0b10 [27] was employed for selection of the best-fit nucleotide substitution model for the phylogenetic analysis. The sequence alignment was analysed using Maximum Likelihood with the Tamura-Nei (TrN) model of evolution [28] with gamma distribution and the proportion of invariable sites (TRN + Γ + I) estimated from the data set using PhyML 2.4.4 [29]. The robustness of the Maximum Likelihood tree was evaluated by the bootstrapping method with 100 replicates using PhyML. In addition, a phylogram was constructed using the neighbour-joining [30] method with the Jin-Nei model using the PHYLIP package [31] within BioManager. The neighbour-joining tree was assessed by bootstrapping 100 replicates. KUNV strain MRM61C (D00246) was used as an outgroup in the construction of the phylogenetic trees.
Nucleotide sequences were translated using the standard genetic code and aligned using CLUSTAL W. A distance matrix was produced to determine the percentage amino acid sequence homology between isolates of MVEV and to investigate the presence of unique amino acid motifs in the amino acid sequences.
Monoclonal antibody (Mab) binding patterns of isolates of MVEV were investigated by inoculation of isolates onto confluent 96-well monolayers of C6/36 cells, incubation for up to 7 days, and subsequent assay by tissue culture enzyme immunoassay [32, 33] using a panel of flavivirus and MVEV-specific Mabs. Mabs used in the phenotypic analysis comprised 4G2, 4G4, ME6, 10C6, 10G4, 2G1, 4F11 and 5E9, all raised to the prototype MVE-1-51 [34–37]. Flavivirus group-reactive Mabs 4G2 (anti-E) or 4G4 (anti-nonstructural (NS) 1 protein) were used as positive controls. Wells that exhibited optical densities of ≥ twice the optical density of uninfected negative control wells and a minimum optical density of 0.2 were deemed positive. Monolayers infected with MVE-1-51 and uninfected C6/36 cell monolayers were used as positive and negative controls, respectively.
Results
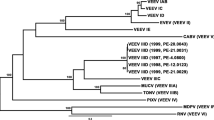
Within the 462 bp nucleotide sequence alignment, 341 (74%) nucleotides were constant, 89 (19%) nucleotides were parsimony informative and 32 (7%) nucleotides represented unique singletons. Phylogenetic trees constructed using the neighbour-joining and maximum likelihood methods shared similar topography. The Maximum Likelihood tree differs from the neighbour-joining tree only in the bootstrap support value and only the Maximum Likelihood tree is presented (Fig. 2). Both methods strongly support two distinct lineages (I and II). Two remaining isolates from PNG did not cluster with either lineage I or II and represent two additional divergent lineages.

Maximum Likelihood phylogenetic tree representing the nucleotide sequence identity of the envelope region (partial) of isolates of Murray Valley encephalitis virus. Bootstrap values from 100 replicates of the Maximum Likelihood/Neighbour-joining phylogenetic trees are located at branch origins
A minimum nucleotide sequence identity of 84.6% (translating to 94.4% amino acid sequence homology) was observed between all isolates of MVEV between 1951 and 2003. Strains of MVEV from WA were present in lineages I and II, and the majority of WA isolates were in lineage I. A nucleotide sequence identity of 96.1% (translating to 97.6% amino acid sequence homology) was observed between all WA strains in lineage I. The minimum nucleotide sequence identity and amino acid homology between all isolates in lineage I, including the prototype MVE-1-51 and two isolates from PNG in 1998 (PNG6523 and PNG6910) [13], was 94.2% and 97.6%, respectively. In general, recent isolates of MVEV from WA and Queensland were more closely related to each other than to older isolates, and isolates were not clustered based on geographical origin. Lineage II consisted of five aberrant strains of MVEV from WA, first isolated in 1973 [38] and most recently in 1995. Isolates in lineage II shared 97.2% nucleotide sequence identity (translating to 99.2% amino acid sequence homology) with each other, and were more closely related to the lineages III and IV from PNG (sharing a minimum of 87.0% nucleotide sequence identity and 96.0% amino acid similarity) than to virus strains in lineage I (with a minimum of 84.6% nucleotide sequence identity and 94.4% amino acid homology). Lineages III and IV comprised two isolates of MVEV from PNG in 1956 (NG156) and 1966 (MK6684) [14], respectively, sharing 91.3% nucleotide sequence identity and 99.2% amino acid sequence homology. The two recent isolates of MVEV from PNG in 1998 were more closely related to Australian isolates of MVEV in lineage I (with a minimum of 94.2% nucleotide sequence identity and 97.6% amino acid homology) than to the older isolates from PNG (sharing 89.6% nucleotide sequence similarity and 96.8% amino acid homology). Isolates of MVEV from western Cape York Peninsula [11] shared high levels of nucleotide sequence identity and 100% amino acid homology with recent isolates of MVEV from WA. CY1189 (from Pormpuraaw, western Cape York Peninsula, 1999) was identical to isolates from Exmouth (P6311) in 1999 and Newman (P6461) in 2000.
Four amino acid substitutions in the partial E gene sequence were unique to lineage II isolates compared to the MVE-1-51 prototype: E-205 (thr to ser), E-232 (glu to asp), E-275 (ser to thr) and E-276 (ser to gly). In addition, two amino acid substitutions were found only in isolates belonging to lineages II, III and IV: E-229 (ala to ser) and E-240 (val to met). All Australian isolates after 1951 had an amino acid substitution at E-238 (ile to val) compared to MVE-1-51. The only non-conservative change was the substitution of ala with ser at E-229 in lineages II, III and IV viruses.
A previous unpublished study that examined the Mab binding profiles of a small panel of MVEV isolates from Australia and PNG using a similar assay to that used in this study indicated that some strains of MVEV, including the PNG isolates NG156 and MK6684 and North Western Australian isolate OR156, could be distinguished antigenically [35]. To assess whether the different lineages of MVEV identified in our study also displayed distinct antigenic patterns, binding profiles of a panel of MVEV-specific antibodies were assessed to each isolate in ELISA. This analysis revealed that three Mabs (10G4, ME6 and 4F11) showed differential binding to MVEV isolates, separating the panel into five groups (Table 2). Mab 10G4 recognised the majority of MVEV isolates tested, but failed to bind to all isolates in lineages II, III and IV, which contained the early PNG isolates and OR156-like isolates, and two isolates in lineage I (K4629 and CY2692). In contrast, Mab ME6 recognised all isolates of lineages II, III and IV, but failed to recognise 12 of the 30 isolates in lineage 1. Notably all the viruses that lacked the ME6 epitope clustered together in the tree, and tended to be more recent isolates. Mab 4F11 recognised all isolates except the 1956 human isolate from PNG (NG156).
Discussion
This study revealed that recent strains of MVEV, isolated from geographically separated areas of WA at the same time, are closely related and suggests that strains of MVEV active at any one time probably originate from a common gene pool. The results provide further evidence that MVEV moves large distances from one area to another, confirming those of other researchers [23, 39–42] who hypothesised that MVEV moves from northern regions where the virus is enzootic, to other areas further south under the influence of certain climatic conditions. The mechanism by which MVEV moves such large distances is not known, although it is possible that viraemic migratory waterbirds [39, 43] or wind-borne infected mosquitoes [44, 45] are involved.
This study extends previous investigations of the relationship between isolates of MVEV from disparate areas of WA by almost 20 years [41, 46, 47]. The study showed that only minor genetic changes occur over time and are not related to geographic origin, similar to findings by previous researchers [23, 41, 42, 46, 47]. The maximum nucleotide sequence divergence between the majority of Australian isolates of MVEV between 1951 and 2003 in 462 bases of the E region was 3.9%, slightly greater than the approximate 2% divergence observed between Australian isolates from 1951 to 1984 [47]. However, the rate of change between 1951 and 2003 (0.07% per year) is similar to the rate of change between 1951 and 1984 (0.06% per year), which suggests a relatively constant rate of nucleotide change over time.
Interestingly, in this study four strains of MVEV from Kununurra in the northeast Kimberley region of WA were genetically related to a unique variant of MVEV, OR156, initially described by Lawson [47] and Coelen and Mackenzie [46] following partial nucleotide sequence analysis, restriction enzyme mapping and RNase T1 oligonucleotide fingerprinting. The origin of OR156 was unknown, although it was suggested that the source might have been islands to the north of Australia, such as PNG or the Indonesian Archipelago [1, 41, 46, 47]. The closer relationship of the OR156 lineage II to old lineage III and IV PNG isolates of MVEV than to the predominant Australian lineage I supports the hypothesis that the variant form of MVEV was introduced from outside Australia. It was generally thought that the OR156 lineage had not become established. However, detection of additional representatives of the same unique genetic lineage over an 18-year period indicates that either lineage II is enzootic with intermittent activity, or that there have been repeated introductions.
Lineage II has not been detected outside the northeast Kimberley region of WA, even though environmental conditions were apparently conducive to movement of MVEV from enzootic regions in the northeast Kimberley region to areas further south in 1991 and 1995 (2 years when lineage II was detected at Kununurra), as evidenced by seroconversions in sentinel chicken flocks (A. Broom, unpublished results). Mosquito collections for virus detection in the Pilbara and more southerly regions of WA were undertaken less regularly than the annual surveys conducted in the Kimberley region and it is possible that more comprehensive surveys would have led to the detection of lineage II MVEV in mosquitoes from more southerly regions, if it had moved beyond Kununurra. Alternatively, lineage II may only exist in transmission cycles at Kununurra, which would suggest that this lineage is evolving independently, is possibly restricted to a unique ecological niche, and is unable to move to other areas due to the presence of an as yet undefined physical or biological barrier. Given that the more predominant strains of MVEV appear to move readily between the northeast Kimberley and more distant regions, this barrier is unlikely to be a physical one, and is more likely to be related to fundamental ecological or biological characteristics of lineage II viruses. Isolates of lineage II and the more predominant Australian MVEV lineage I were obtained from Cx. annulirostris and Cx. pullus, indicating that the likely vectors and their associated host-feeding preferences may be the same. However, unique monoclonal antibody-binding patterns for lineage II viruses is evidence that this lineage is phenotypically distinctive. In addition, McMinn et al. [22] demonstrated that amino acid substitutions in certain parts of the E protein of MVEV, particularly between E-270-277 (referred to as the hinge region), were often associated with phenotypic changes including increased sensitivity to neutralisation, reduced haemagglutination activity, attenuation of neuroinvasiveness and altered growth kinetics. In this study, amino acid substitutions found in lineage II viruses were mostly conservative and, unlike those described by McMinn et al. [22], may not be biologically significant. It must also be acknowledged that mutations potentially introduced into the genome following multiple passages through mice and cell cultures during the virus isolation process may be responsible for some differences. However, there was no significant correlation between isolate passage history and the number of amino acid substitutions (r = −0.188, P = 0.295) (SigmaStat for Windows version 3.5). Furthermore, the passage history of MVEV isolates used in this study is similar to those used in other phylogenetic and phenotypic studies [16, 18, 19]. This indicates that passage history is not associated with the major genetic and antigenic relationships between the MVEV lineages described here. Although the significance of the amino acid sequence differences observed in this study is not known, it is possible that some of these and/or other as yet undescribed amino acid changes may have lead to alterations in biological characteristics such as replication fitness, host specificity and vector competence, and may explain why lineage II viruses appear to be restricted to a small geographic location. Indeed, a previous study by Lawson [47] found differences in replication efficiency and virulence for OR156 compared with other more virulent isolates of MVEV. Further nucleotide sequence analyses, and mutagenesis and virulence studies using representative strains of each lineage of MVEV are underway to elucidate biological differences between the two distinct lineages found in Australia. In addition, the co-circulation of different lineages of MVEV at one locality provides a unique opportunity to investigate differences in virus ecology, epidemiology and transmission.
Recent lineage I isolates of MVEV from Western Province in PNG in 1998 [13] were more closely related to mainland Australian lineage I strains than older lineages III and IV isolates of MVEV from PNG [23, 42]. It has been hypothesised that the highlands in PNG may be a barrier to virus movement southwards into Australia [23]. While the highlands may indeed be a physical barrier, the close genetic relationship between isolates of Japanese encephalitis virus from Western Province of PNG and Australia provides circumstantial evidence that, south of the highlands, certain flaviviruses may move from PNG to Australia, possibly via wind-blown infected mosquitoes [13, 48] or viraemic animal hosts. Likewise, the close relationship between isolates of MVEV from Balimo in Western Province of PNG, south of the highlands, may be evidence for virus movement from northern Australia into PNG via similar mechanisms.
Consistent with a previous unpublished report by one of the authors [35], we also demonstrated that the older lineages III and IV isolates of MVEV from PNG and lineage II isolates from Kununurra were antigenically distinct from lineage I viruses (with the exception of CY2692). Additional antigenic analysis of the Australian MVEV isolates in lineage I revealed that 22 could be further separated into two groups that strongly correlated with time but not place of isolation (Table 2). Ten isolates from various locations prior to 1985, including the 1951 prototype virus to which the Mabs were prepared, fell into phenotypic Group 1 (Table 2) and retained all the NS1 epitopes recognised by the panel of six MVEV-specific Mabs. Twelve viruses (in Groups 3 and 4) that were isolated after 1988 had lost the epitope recognised by Mab ME6. Some discrepancies between the phylogenetic and phenotypic analyses were observed. However it should be noted that all Mabs used in this analysis react with conformational epitopes and that a single nucleotide change in the viral genome can potentially alter the binding site for each Mab. Nevertheless, despite the few exceptions, these data support the conclusions from the sequence analysis, and provide phenotypic evidence of a gradual temporal evolution amongst Australian MVEV isolates that is not associated with place of isolation. The biological importance of the amino acid differences and altered Mab binding epitopes observed in this study is the subject of further study.
References
J.S. Mackenzie, M.D. Lindsay, R.J. Coelen, A.K. Broom, R.A. Hall, D.W. Smith, Arch. Virol. 136, 447 (1994)
I.D. Marshall, in The Arboviruses: Epidemiology and Ecology, ed. by T.P. Monath (CRC Press, Boca Raton, 1988), p. 151
A.K. Broom, M.D.A. Lindsay, S.A. Harrington, D.W. Smith, Vector Borne Zoonotic Dis. 2, 87 (2002)
S.P. Cordova, D.W. Smith, A.K. Broom, M.D. Lindsay, G.K. Dowse, M.Y. Beers, Commun. Dis. Intell. 24, 368 (2000)
J.S. Mackenzie, D.W. Smith, A.K. Broom, M.R. Bucens, Medical J. Aust. 158, 591 (1993)
A.K. Broom, M.D.A. Lindsay, A.J. Plant, A.E. Wright, R.J. Condon, J.S. Mackenzie, Am. J. Trop. Med. Hyg. 67, 319 (2002)
R.A. Hall, A.K. Broom, A.C. Hartnett, M.J. Howard, J.S. Mackenzie, J. Virol. Methods 51, 201 (1995)
A. Broom, K. Sturrock, B. van Heuzen, M. Lindsay, D. Smith, Arbovirus Res. Aust. 8, 43 (2001)
A.K. Broom, M.D.A. Lindsay, A.E. Wright, D.W. Smith, J.S. Mackenzie, Am. J. Trop. Med. Hyg. 69, 277 (2003)
H.F. Chapman, B.H. Kay, S.A. Ritchie, A. van den Hurk, J.M. Hughes, J. Med. Entomol. 37, 736 (2000)
C.A. Johansen, D.J. Nisbet, P. Zborowski, A.F. van den Hurk, S.A. Ritchie, J.S. Mackenzie, J. Am. Mosq. Control Assoc. 19, 392 (2003)
E.L. French Med. J. Aust. 1, 100 (1952)
C.A. Johansen, A.F. van den Hurk, S.A. Ritchie, P. Zborowski, D.J. Nisbet, R. Paru, M.J. Bockarie, J. Macdonald, A.C. Drew, T.I. Khromykh, J.S. Mackenzie, Am. J. Trop. Med. Hyg. 62, 631 (2000)
G.M. Woodroofe, I.D. Marshall Arboviruses from the Sepik district of New Guinea. Canberra: John Curtin School of Medical Research Annual Report, (Australian National University, 1971), pp. 90–91
A.T. Pyke, D.T. Williams, D.J. Nisbet, A.F. van den Hurk, C.T. Taylor, C.A. Johansen, J. Macdonald, R.A. Hall, R.J. Simmons, R.J.V. Mason, J.M. Lee, S.A. Ritchie, G.A. Smith, J.S. Mackenzie, Am. J. Trop. Med. Hyg. 65, 747 (2001)
J.H. Scherret, M. Poidinger, J.S. Mackenzie, A.K. Broom, V. Deubel, W.I. Lipkin, T. Briese, E.A. Gould, R.A. Hall, Emerg. Infect. Dis. 7, 697 (2001)
D.T. Williams, L.F. Wang, P.W. Daniels, J.S. Mackenzie, J. Gen. Virol. 81, 2471 (2000)
L.D. Kramer, L.J. Chandler, Arch. Virol. 146, 2431 (2001)
F.J. May, M. Lobigs, E. Lee, D.J. Gendle, J.S. Mackenzie, A.K. Broom, J.V. Conlan, R.A. Hall, J. Gen. Virol. 87, 329 (2006)
L. Dalgarno, D.W. Trent, H.H. Strauss, C.M. Rice, J. Mol. Biol. 187, 309 (1986)
J.T. Roehrig, Adv. Virus Res. 59, 141 (2003)
P.C. McMinn, E. Lee, S. Hartley, J.T. Roehrig, L. Dalgarno, R.C. Weir, Virology 211, 10 (1995)
M. Lobigs, I.D. Marshall, R.C. Weir, L. Dalgarno, Virology 165, 245 (1988)
J.D. Thompson, D.G. Higgins, T.J. Gibson, Nucleic Acids Res. 22, 4673 (1994)
S. Kumar, K. Tamura, M. Nei, Brief Bioinformatics 5, 150 (2004)
D. Posada, K.A. Crandall, Bioinformatics 14, 817 (1998)
D.L. Swofford PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4. Sinauer Associates, Sunderland, Massachusetts, (2005)
K. Tamura, M. Nei, Mol. Biol. Evol. 10, 512 (1993)
S. Guindon, O. Gascuel, Syst. Biol. 52, 696 (2003)
N. Saitou, M. Nai, Mol. Biol. Evol. 4, 406 (1987)
J. Felsenstein, PHYLIP (Phylogeny Inference Package) Version 3.5c. Department of Genetics, University of Washington, Seattle. (1993)
A.K. Broom, R.A. Hall, C.A. Johansen, N. Oliveira, M.A. Howard, M.D. Lindsay, B.H. Kay, J.S. Mackenzie, Pathology 30, 286 (1998)
S.C. Adams, A.K. Broom, L.M. Sammels, A.C. Hartnett, M.J. Howard, R.J. Coelen, J.S. Mackenzie, R.A. Hall, Virology 206, 49 (1995)
R.A. Hall, G.W. Burgess, B.H. Kay, Immunol. Cell Biol. 66, 51 (1988)
R.A. Hall, PhD Thesis. James Cook University, Townsville, Queensland, (1989)
E.A. Henchal, M.K. Gentry, J.M. McCown, W.E. Brandt, Am. J. Trop. Med. Hyg. 31, 830 (1982)
D.C. Clark, M. Lobigs, E. Lee, M.J. Howard, K. Clark, B.J. Blitvich, and R.A. Hall, J. Gen. Virol. 88, 1175 (2007)
C.G. Liehne, S. Leivers, N.F. Stanley, M.P. Alpers, S. Paul, P.F.S. Liehne, K.H. Chan, Aust. J. Exp. Biol. Med. Sci. 54, 499 (1976)
J.A. Forbes, Murray Valley encephalitis 1974—also the epidemic variance since 1914 and predisposing rainfall patterns. (Australian Medical Publishing Co., Glebe, 1978)
N. Nicholls, Aust. J. Exp. Biol. Med. Sci. 64, 587 (1986)
R.J. Coelen, M.A. Lawson, L.M. Flynn, J.S. Mackenzie, Arbovirus Res. Aust. 5, 55 (1989)
M. Lobigs, I.D. Marshall, R.C. Weir, L. Dalgarno, Aust. J. Exp. Biol. Med. Sci. 64, 571 (1986)
J.A.R. Miles, D.W. Howes, Med. J. Aust. 1, 7 (1953)
B.H. Kay, R.A. Farrow, J. Med. Entomol. 37, 797 (2000)
S.A. Ritchie, W. Rochester, Emerg. Infect. Dis. 7, 900 (2001)
R.J. Coelen, J.S. Mackenzie, J. Gen. Virol. 69, 1903 (1988)
M.A. Lawson, PhD Thesis. The University of Western Australia, Nedlands, Perth, Western Australia (1988)
J.S. Mackenzie, M. Poidinger, D. Phillips, C. Johansen, R.A. Hall, J. Hanna, S. Ritchie, J. Shield, and R. Graham, in Factors in the emergence of arbovirus diseases. Ed. by J.F. Saluzzo, B. Dodet (Elsevier, Paris, 1997), pp. 191–136
Acknowledgments
The authors wish to acknowledge the assistance of Dr. Michael Lindsay, Susan Harrington, Adrian Stratico and Tony Wright (WA Department of Health), Brenda van Heuzen, Steven Crocker, Rosa Duthie, Maria Beilin, Margaret Wallace and Keryn Sturrock (The University of WA), Debra Nisbet and Dr. Andrew van den Hurk (The University of Queensland) and Dr. Scott Ritchie (Queensland Health) with mosquito collections and virus isolations. We also thank James Conlan and Dr. Helle Bielefeldt-Ohmann (The University of Queensland) for assistance with sequencing the isolates NG156 and MK6684. The late Professor Neville Stanley and Peter Liehne initiated and obtained funding for the early studies on MVEV in WA. We also thank Dr. Malcom Lawson (The University of WA) for helpful scientific discussions. This research was funded by the WA Department of Health.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Johansen, C.A., Susai, V., Hall, R.A. et al. Genetic and phenotypic differences between isolates of Murray Valley encephalitis virus in Western Australia, 1972–2003. Virus Genes 35, 147–154 (2007). https://doi.org/10.1007/s11262-007-0091-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-007-0091-2