
Abstract
This study provides the first comprehensive report on the molecular characteristics of African swine fever virus (ASFV) variants in Serbia between 2019 and 2022. Since its first observation in July 2019, the disease has been found in wild boar and domestic swine. The study involved the analysis of 95 ASFV-positive samples collected from 12 infected administrative districts in Serbia. Partial four genomic regions were genetically characterized, including B646L, E183L, B602L, and the intergenic region (IGR) between the I73R-I329L genes. The results of the study suggest that multiple ASFV strains belonging to genotype II are circulating in Serbia, as evidenced by the analysis of the IGR between I73R-I329L genes that showed the most differences. Furthermore, the phylogenetic analysis of the B602L gene showed three different clades within the CVR I group of ASFV strains. Regarding the IGR, 98.4% were grouped into IGR II, with only one positive sample grouped into the IGR III group. These findings provide essential insights into the molecular characteristics of ASFV variants in Serbia and contribute to the knowledge of circulating strains of ASFV in Europe. However, further research is necessary to gain a better understanding of ASFV spread and evolution.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
African swine fever (ASF) represents one of the most significant diseases affecting the swine industry. The disease is caused by the African swine fever virus (ASFV), classified within the genus Asfivirus, of the family Asfaviridae (Alonso et al. 2018). The viral genome is composed of a large double-stranded DNA (170–190 kb), surrounded by an icosahedral capsid (1892 to 2172 capsomers) and an outer lipid layer, with the size of the virion around 175–215 nm in diameter (Alonso et al. 2018). The genome of ASFV has 150 to 189 open reading frames (ORF), and the number, as well as the length of the genome, varies depending on the genotype (Dixon et al. 2013). ASFV persists in wildlife hosts such as warthogs, bushpigs, and soft ticks of Ornithodorus spp. However, if domestic pigs or wild boars are infected with ASFV, it leads often to fatal disease, although wild boar survivors from moderate virulent strains have been detected (Nurmoja et al. 2017; Gallardo et al. 2018). In Africa, the virus is maintained within the sylvatic cycle between warthogs and soft O. spp. ticks as well as domestic pigs where a high genetic diversity of 24 genotypes was detected (Bastos et al. 2003; Quembo et al. 2017). Only genotypes I and II of ASFV have been recorded outside of the African continent. Between 1957 and 1993, there were several reports on ASFV genotype I incursions into Europe and the Americas. Although almost all of these outbreaks were successfully controlled and eradicated from the infected countries outside Africa, except Sardinia. Enzootically, genotype I has been present in wild boar and domestic pig populations in Sardinia for over four decades and was recorded for the first time in 2021 in China during an outbreak in domestic pigs (Sun et al. 2021). ASF became prominent when the disease emerged from the African continent in 2007, initially infecting domestic pigs in Georgia (Rowlands et al. 2008). The virus was then transmitted to Russia (Gogin et al. 2013), and further into Europe, and recently it has been reported in Oceania and Central America (Jean-Pierre et al. 2022; Department for Environment 2023). Although wild boar remains the main reservoir of ASFV in Eurasia within the wild boar-habitat cycle (Sauter-Louis et al. 2021a; Denstedt et al. 2021; Cadenas-Fernández et al. 2022), there are different possible O spp. vectors (O. erraticus, O. verrucosus), besides O. moubata, on the European continent (South-Western Europe, and Eastern Europe), which could contribute to the spread of the virus (European Centre for Disease Prevention and Control (ECDC) 2018; de Oliveira et al. 2019; Pereira De Oliveira et al. 2020). Due to the low transmission/infectivity rate of ASFV, coupled with the habitat–wild boar transmission cycle, and the emergence of less virulent strains, the virus can remain within the population and not become a self-limiting disease. (Gallardo et al. 2018; Chenais et al. 2019; Sauter-Louis et al. 2021a). Another significant fact is the very high tenacity of ASFV, which enables the virus to survive for long periods at 0–4 °C temperatures, and at a high pH range, thus allowing the ASFV to survive in a wide range of environments, even in processed meat (Mazur-Panasiuk et al. 2019). Such resilience allows the virus to be transported through human activities over long distances and continues to be infectious (Mazur-Panasiuk et al. 2019).
Even though genotype II has been detected in more than 60 different countries on different continents since 2007, ASFV continues to be very stable. In order to track different variants of the virus as it passes through the susceptible population certain genetic markers should be sequenced. By sequencing other regions of the genome, such as the E183L (Gallardo et al. 2009), B602L genes (Gallardo et al. 2014), or other genes with a higher mutation frequency, a higher sequencing resolution can be achieved necessary for tracking different viral strains (Mazloum et al. 2023a). The 3’ end of the B646L gene (encoding the p72 protein) can be used to classify ASFV into 24 genotypes (Boshoff et al. 2007; Quembo et al. 2016, 2017; Achenbach et al. 2017). This classification is an essential tool for tracking the virus as it appears in new territories, as was done when the virus first emerged in Georgia (Ramishvili 2007).
ASFV was introduced in Serbia in July and August 2019, probably by humans, from neighbouring infected countries (Milićević et al. 2019). The first two outbreaks were in domestic pigs, and immediately the stamping-out policy was implemented. In January 2020, the disease was detected for the first time in wild boar and has been present since then in the wild boar population in infected areas in Serbia (Nešković et al. 2021; Glišić et al. 2023; Prodanov-Radulović et al. 2023). ASFV has been localised within the eastern and southern parts of Serbia, where the highest density wild boar population is inhabited. The critical risk factor for enzootics maintenance is a very high percentage of backyard farms, which enable the circulation of the virus in wild boar and domestic pig populations (Polaček et al. 2021). Based on official data, there were 18 reported outbreaks of ASFV in domestic pigs in Serbia in 2019 (European Commission: Animal Disease Information System 2019). By 2020, the disease had spread to wild boars, resulting in 63 reported outbreaks, with 16 in domestic pigs (European Commission: Animal Disease Information System 2020). The number of outbreaks continued to increase in 2021, with 33 in domestic pigs and 71 in wild boars (European Commission: Animal Disease Information System 2021). In 2022, the highest number of outbreaks since the disease was first reported was recorded, with 107 in domestic pigs and 146 in wild boars (European Commission: Animal Disease Information System 2022).
In the recent study done by Gallardo et al. (2023), the Serbian index case was classified within the genetic group 19 based on the analysis of 6 genomic markers. Since the disease has been present in the country for four years, the genomic characterisation of current ASFV variants should be further investigated. In this study, we aimed to determine the circulating strains of ASFV, by sequencing four different genomic markers and thus give a comprehensive report on the molecular characteristics of the strains currently present in the country and their relationship with the strains circulating in Europe. Such information will help us with disease tracing, and allow us to determine the transmission routes better.
Materials and methods
Samples selection
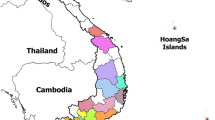
Samples were selected from 280 ASFV-positive wild boar cases and 174 domestic pig outbreaks reported in 12 administrative districts in Serbia from June 2019 to October 2022 (Fig. 1). Samples were stored in the national sample bank and were deliberately chosen for the best space–time coverage. Geographically, each municipality in which ASFV had been recorded was included, as well as wild boar and domestic pig samples from each year when the disease was detected. The samples consisted of tissues (spleen and kidney), blood swabs, and bone marrow extracts from found dead wild boar. Tissue samples were homogenized with a mortar and pestle in a 1:10 ratio with PBS. The swabs were immersed in 1 ml of PBS and further homogenized. Thus, the samples were centrifuged at 1500 × g for 10 min. The supernatants were decanted and stored at -80 °C until further use. Viral DNA was extracted using the IndiSpin Pathogen kit (IndiSpin Pathogen Kit, Indical Bioscience GmbH, Leipzig, Germany) following the manufacturer's instructions.

Map of infected administrative districts in the country. Positive ASF cases were chosen for phylogenetic analysis. Administrative districts where ASFV was detected from 2019 to 2022 are highlighted in yellow
To detect the ASFV genome, the samples were first tested using real-time PCR with primers and a probe targeting the B646L gene which encodes the p72 protein, as described by King et al. (2003). The master mix used included 12 μl of Luna Universal Probe qPCR Master Mix (from New England BioLabs, Ipswich, MA, USA), 1 μl of 10 mM each primer, 0.5 μl of 10 mM probe, and 5 μl of DNA template, and the rest of the mixture was supplemented with RNA-free water to a total volume of 20 μL. The temperature profile involved initial denaturation at 95 °C for 1 min, followed by 50 cycles of 95 °C for 15 s and 60 °C for 30 s.
PCR amplification for sequencing
We selected 95 samples for sequencing based on their Ct values obtained from the real-time PCR. Only samples with Ct values less than 25 were further analysed. We amplified four different genomic markers for phylogenetic analysis. The 3’-end of the B646L gene (encoding the p72 protein) was amplified using the primers described by Bastos et al. (2003), to determine the genotype, while the p54—coding gene E183L was amplified by primers described by Gallardo et al. (2009). For further sequencing, we targeted the central variable region (CVR) located within the B602L gene using primers published by Gallardo et al. (2009) and the tandem repeat sequences (TRS) within the intergenic region (IGR) between the I73R and I329L genes using the primers described by Gallardo et al. (2014). Each reaction consisted of the 10 μl of HotStar Master Mix (Qiagen, Les Ulis, France), 0.6 μl of each 10 mM primer, and 2 μL of DNA template. The remaining volume, up to 20 μl, was filled with RNA-free water. The temperature profiles used in the study involved several steps. First, the samples were initially denatured at 95 °C for 15 min. This was followed by 40 cycles of denaturation at 95 °C for 30 s, annealing at 50 °C for 30 s (for the B646L and E183L genes) or at 55 °C (for the CVR and IGR), extension at 72 °C for 1 min, and a final extension at 72 °C for 10 min. The products were visualized using electrophoresis on a 1.5% agarose gel, with expected lengths of around 500 bp for the B646L gene, around 600 bp for the E183L gene, 500 bp for the CVR, and 370 bp for the IGR. Samples which were deemed positive were further purified using the GeneJET PCR purification kit (ThermoFisher Scientific, Waltham, MA, USA), and sequenced at LGC, Biosearch Technologies, Germany, using the Sanger sequencing method.
Sequence analysis
Using the Geneious Prime software (developed by Dotmatics, Boston, MA, USA), consensus sequences were created by trimming and aligning the sequences of the B646L gene to a length of 297 bp. These sequences were then compared with 41 strains, including all 24 genotypes of ASFV, which were obtained from the National Center for Biotechnology Information (NCBI) (Table S1.). The sequences of the E183L gene were aligned and compared with 21 NCBI sequences (Table S1.), after being trimmed to a length of 534 bp. Similarly, the sequences of the B602L gene were trimmed to a length of 343 bp, aligned, and compared with 46 NCBI sequences (Table S1.). Lastly, the sequences of the IGR located between the I73R and I329L genes were trimmed to a length of 241 bp, aligned, and compared with NCBI sequences (Table S1.). To construct the phylogenetic trees, the Molecular Evolutionary Genetic Analysis (MEGA X) software was utilized. The Neighbour-Joining method, Jukes-Cantor model, and Gamma discrete (5.00) differences were applied to the B646L and E183L genes, with 1000 bootstrap replicates and uniform rates among sites. Meanwhile, the Maximum Likelihood method, Kimura 2 parameter model, and Gamma (5.00) discrete differences were used for the B602L genes, also with 1000 bootstrap replicates and uniform rates among sites. The models used for the phylogenetic analysis were determined based on the outcomes of the MEGAX Find Best DNA/Protein model features.
Results
In total, 95 positive samples with Ct values lower than 25 were selected for the sequencing and phylogenetic analysis. Sequences from all four genomic regions were obtained from 64 samples (Table S1). Good-quality sequences were obtained from 85 samples for the partial B646L gene, 89 for the E183L gene, 78 for the partial B602L gene, and 64 for the IGR.
Phylogenetic analysis of the partial B646L gene
For amplification of the C-terminus end of the B646L gene, 85 (89.5%) good-quality sequences out of the 95 sequenced samples were obtained. The study's sequences were compared with other sequences from the NCBI (refer to Table S1). In addition, the sequences were submitted to the NCBI GenBank, and their respective accession numbers are OQ336124–OQ336192 and OQ060619–OQ060634. The sequences of genotypes I-XXIV were also included. The phylogenetic analysis revealed that all ASFVs from Serbia cluster within genotype II together with the Georgia 2007 reference strain (FR682468.2). (Fig. 2).

The genetic relationships between the partial B646L gene sequences of Serbian strains (OQ336124-OQ336192 and OQ060619-OQ060634) and other strains obtained from the NCBI (see Table S1.) were illustrated using a phylogenetic tree. The analysis was conducted in MEGA X software, employing the Maximum Likelihood Method and the Jukes-Cantor model, with 1000 bootstrap replicates and discrete Gamma distribution (+ G) with 5 rate categories. Additionally, a fraction of sites that are evolutionarily invariable (+ I) was assumed. To simplify the tree, branches corresponding to partitions reproduced in less than 70% of bootstrap replicates were collapsed. The phylogenetic analysis included all 24 genotypes (see Table S1.). The best-fit model was determined using the Find Best DNA/Protein Models feature of the Mega X software. Serbian samples were marked with black dots on the tree
Phylogenetic analysis of the E183L gene
Out of 95 sequenced strains, 89 (93.7%) good-quality sequences were obtained. Sequences examined in the study (OQ335972-OQ336061) were compared to the corresponding sequences from the NCBI (Table S1.). Sequences AY261360, AY261362, AY261363, AY261364, AY261366, FJ174420, FJ174421, FJ174422, FJ174425, FJ174430, FR682468, and X84889, were added as sequences representing different subgenotypes that can be differentiated by sequencing the E183L gene, and represent out-of-group sequences of genotypes I, V, VIII, X. The phylogenetic analysis revealed no discernable mutations between the sequences in this study, and all Serbian ASFVs clustered within the genotype II group (Fig. 3).

A phylogenetic tree was created to show the genetic relationships between the Serbian E183L gene sequences (OQ335972—OQ336061) and the sequences obtained from NCBI (Table S1.). The analysis was done in MEGA X software using the Maximum Likelihood method and Jukes-Cantor model. The analysis included 1000 bootstrap replicates and a uniform rate among site Gamma distribution (+ G) with 5 rate categories. It was also assumed that some sites are evolutionarily invariable (+ I). Branches that were reproduced in less than 70% of bootstrap replicates were collapsed. The Mega X software's "Find best DNA/Protein Models" feature was used to determine the best-fit model. The Serbian samples are represented by sequences marked with black dots
Phylogenetic analysis of the CVR within the B602L gene
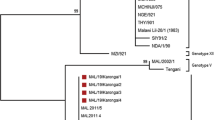
Out of the 95 samples, 76 sequences (80%) with good quality were obtained through CVR amplification of the B602L gene. These sequences were submitted to the NCBI GenBank and assigned accession numbers OQ336062-OQ336123 and OQ060635-OQ060650. The CVR sequences obtained from the Serbian samples were compared to 104 sequences from genotype II ASFVs that were retrieved from the NCBI (Table S1.). During the phylogenetic analysis, it was observed that when the Serbian variants were compared to the partial B602L gene sequences from the NCBI, there were seven distinct clades (Fig. 4.). Serbian sequences split into four different clades, the majority of strains clustering together with the ASFV Georgia 2007/1 reference strain (Clade IV). Variant SRB/2021/5431 split into a separate clade (Clade II) while other Serbian sequences which exhibited a T/A change at the 318th position of the complete gene, split together into Clade III. Also, three Serbian strains split into clade IV. Variant SRB/2020/7511 clustered together with other Serbian strains which exhibited the T/A 318 SNP into Clade III. CVR SNP mutations were not observed in the samples collected in 2019 because the cases were imported from affected countries through human activities.

A phylogenetic tree was created to show the genetic relationships between Serbian B602L gene sequences (OQ336062-OQ336123 and OQ060635- OQ060650) and sequences obtained from NCBI (Table S1.). The analysis was conducted using the Maximum Likelihood Method and Jukes-Cantor model in MEGA X software. The analysis included 1000 bootstrap replicates and a uniform rate among site Gamma distribution (+ G) with 5 rate categories. It was also assumed that some sites are evolutionarily invariable (+ I). Branches that were reproduced in less than 70% of bootstrap replicates were collapsed. The best-fit model was calculated using the "Find best DNA/Protein Models" feature of the Mega X software. The sequences from Serbia are marked with black dots
SNP recorded within the CVR of the partial B602L gene
Out of 76 ASFV strains from Serbia, 16 showed a synonymous mutation B602L T/A mutation at the 318th position of the complete B602L gene. This means that 21.1% of the sequences had this specific mutation. Three nucleotide changes were found in the SRB/2020/7511 variant. One of them was the synonymous T/A change at the 318th position the other change was a nonsynonymous G/A change which occurred at the 552nd position, resulting in an amino acid change from methionine to isoleucine at position 184th of the B602L protein sequence. The third change was a synonymous T/A change which occurred at the 583rd position, resulting in a change from cysteine to serine at position 195th of the B602L amino acid sequence. These changes are shown in Fig. 5. The substitution of methionine with isoleucine, and cysteine with serine in the SRB/2020/7511 variant does not result in any change in the polarity of the amino acid side chain. Regarding the SRB/2021/5431 variant, two changes were observed—a synonymous T/A nt change at the 572nd position and a nonsynonymous T/C nt change at the 642nd position, which resulted in the substitution of aspartic acid with valine at the 191st position in the complete amino acid sequence. The substitution of the amino acid aspartic acid with valine in the SRB/2021/5431 variant results in a shift in the polarity of the amino acid side chain. The other 60 strains exhibited a complete match in similarity to the ASFV Georgia 2007/1 reference strain (FR682468.2).

A) Nucleotide changes observed within the CVR of the partial B602L gene. The sequences were compared with the ASFV Georgia 2007/1 reference strain. Different groups were formed based on the changes observed within the partial sequences of the B602L gene. In total eight different groups have been highlighted in different colours. b) Amino-acid changes observed within the CVR of the partial B602L protein sequence as compared with the ASFV Georgia 2007/1 reference strain
The phylogenetic analysis of the IGR between I73R and I329L genes
Out of 95 sequenced samples, 64 (67.4%) good-quality IGR sequences were obtained and submitted to the NCBI GenBank under accession number OQ336193—OQ336256. We compared the IGR sequences obtained from the 95 samples with homologous sequences from the NCBI database (Table S1.). After analyzing the IGR sequences obtained from the 95 samples, we identified two distinct variants that are currently circulating in Serbia. Nearly all ASFVs from Serbia (98.4%) have three “TATATAGGAA”, indicating the presence of the IGR II variant, which is the most common variant in Europe. One ASFV from Serbia (SRB/2022/777) (GenBank Ac Nº OQ336250) had four “TATATAGGAA” repeat segments and was identified as an IGR III variant (Fig. 6, Table S1.). This variant has been previously described in viruses from Poland, China, Vietnam and South Korea. Two different SNPs were identified within the IGR-II variant. Seven strains had a thymidine insertion at the 245th position of the IGR, while six sequences showed a C/A SNP at the 56th position of the IGR. These SNPs have not been previously recorded in other genotypes II ASFVs.

Alignment of four IGR variants based on the number of “TATATAGGAA” repeat segments. The IGR IV variant is represented by the Pol19_01529_C8/19, and Pol19_30156_O10/19 variants from Poland, the IGR III variants are represented by the China/Guangxi/2019/domestic pig, and the Serbian SRB/2022/777 variants. The IGR II variants displayed are the SRB/2021/12220, SRB/2021/12229, SRB/2021/1238, SRB/2020/177, SRB/2021/2437 SRB/2022/5844, SRB/2019/6022, SRB/2022/6719, SRB/2022/6203, SRB/2022/6896, SRB/2021/3391, SRB/2021/2347, SRB/2020/4435, Ukr12/Zapo, Belgium/2018/Etalle variants. The IGR I variants are represented by the ASFV Georgia 2007/1, China/Jilin/2018/boar and the Irkutsk2017
Discussion
The disease gained prominence after 2007, when the African swine fever virus (ASFV) spread through the Caucasus into Russia and then west into Europe. Since then, it has been reported in many European countries, including Armenia, Azerbaijan, Russia, Ukraine, Moldova, Belarus, Poland, Romania, Slovakia, Germany, Greece, Hungary, Italy, Belgium, Bulgaria, the Czech Republic, Estonia, Latvia, Lithuania, Serbia, and North Macedonia, and ASF remains the greatest threat to swine production, according to (Rowlands et al. 2008; Oļševskis et al. 2016; Koeltz et al. 2018; Sargsyan et al. 2018; Milićević et al. 2019; Sauter-Louis et al. 2021b; Woźniakowski et al. 2021; Jo and Gortázar 2021; Iscaro et al. 2022; Gallardo et al. 2023; Ladoși et al. 2023). Only two countries have managed to successfully eradicate ASF in wild boar, Belgium and the Czech Republic, with the latter having a reemergence event at the end of 2022 as reported by World Organization for Animal Health (WOAH) (WOAH 2022).
ASFV is one of the most complex animal viruses, with a large genome of 170–193 kb and a number of substitutions per nucleotide per year at a rate of 10–4 (Faburay 2022), which is considered high for dsDNA viruses and a usual range for most RNA viruses (Biek et al. 2015). Although different regions of the virus mutate at different rates, the size of the virus genome makes tracing such mutations difficult.
According to Bastos et al. (2003) partial sequencing of the C-terminal end of the B646L gene, which encodes the p72 protein, is necessary to detect and determine the genotype. The genotypes exhibit high homogeneity, with only certain subgenotypes, such as I (a, b, c, d), V (a, b), X (a, b), and XX (a, b), showing differentiation. Differentiation of these subgenotypes can be achieved through sequencing of the E183L gene, which encodes the p54 protein, as noted by Gallardo et al. (2009). However, sequencing only the B646L gene and the E183L gene may not provide sufficient resolution for tracing the epidemiology of genotype II of ASFV (Faburay 2022). The majority of the ASFV genome is well conserved, showing little to no variation between strains.
To achieve a more detailed sequencing and tracing of different ASFV strains, various genomic markers need to be examined. In recent studies, a sequencing method that utilizes multiple genes has been described. This method can distinguish European ASFV-II genotypes into 25 distinct groups by sequencing six independent genomic regions of ASFV (Gallardo et al. 2023; Mazloum et al. 2023b). The CVR, which contains tandem repeats of amino acids, enables further differentiation of ASFV strains (Nix et al. 2006). By analyzing the IGR between the I73R and I329L genes, genotype II of ASFV can be divided into four variants that have been reported in both Europe and Asia (Gallardo et al. 2014). By analyzing the TRS within the IGR between the 9R-10R genes of MGF505 or that present in the O174L gene, differentiation into eight and two variants can be achieved, respectively. Partial sequencing of the K145R and I215L genes enables differentiation into three variants based on the SNPs (Mazur-Panasiuk et al. 2020; Gallardo et al. 2023; Mazloum et al. 2023b).
Next-generation sequencing (NGS) does allow for higher-resolution sequencing, although such methods remain laborious and difficult to use while offering little epidemiological information (Forth et al. 2020). Although Forth et al. (2023) managed to conduct successful epidemiological tracing of ASF strains through a combination of NGS and Sanger sequencing methods. Besides mutations, the ASFV genome is characterised by frequent recombination events which occur only at “recombination hotspots”(Li et al. 2020), which represent high-diversity regions such as those harboring TRS (de la Vega et al. 1994; Bao et al. 2022). On the other hand, regions such as the B646L and E183L gene are very low diversity regions, with none too few mutations recorded (Li et al. 2020). Concerning the B646L and the E183L gene, in the present study, all examined samples showed 100% identity with the Georgia 2007 reference strain (FR682468.2), and no mutation or discernable differences were observed. This is in accordance with other reports from ASFV-affected European countries (Rowlands et al. 2008; Gogin et al. 2013; Oļševskis et al. 2016; Pikalo et al. 2020; Sauter-Louis et al. 2021a).
The approach to this study concerns the analysis of tandem repeats found within the CVR and the I73R-I329L genes. The CVR is characterised by the insertion of amino acid tetramers, such as (CADT, NVDT, CASM) n, where n represents the number of times the amino acid sequence is repeated. Different strains of ASFV may have different numbers of repetitions, leading to variations in the CVR (Nix et al. 2006). Variations in the CVR have been used to identify 31 different strains of ASFV, enabling in-depth tracking of local strains in Africa (Gallardo et al. 2009). However, only three CVR groups (Groups I, II, and III) have been observed within the Eurasian genotype II (Gallardo et al. 2023) limiting the utility of this approach in certain contexts. Furthermore, it is possible to obtain further differentiation within genotype II by analysing single nucleotide polymorphisms (SNPs) within the CVR, as demonstrated in strains from Estonia (Vilem et al. 2020), Poland, Lithuania (Gallardo et al. 2023), China (Shi et al. 2022) and the Russian Federation (Mazloum et al. 2022). SNP mutations recorded within the CVR region occur sporadically, and their effect on virus survivability should be further investigated.
In our study, 21.1% of sequenced Serbian ASFVs have a synonymous T/A substitution at the 318 position of the complete B602L gene. Despite that the T/A synonymous substitution at the 318 position of the B602L gene in both wild boar and domestic pigs in samples from 2020–2022, no clustering of strains with this change has been observed. The variants SRB/2020/7511 and SRB/2021/5431 showed two different SNP changes, resulting in an amino acid change. This information can be used to categorize strains within the CVR I group. Both variants were detected in domestic pig outbreaks, with variant SRB/2020/7511 identified in 2020 and variant SRB/2021/5431 identified in 2021. The implementation of a stamping-out policy to quickly resolve domestic pig outbreaks may explain the limited spread of these variants. The majority of variants examined in this study had no changes when compared to the ASFV Georgia 2007/1 reference strain.
A significant difference has been observed between the number of sequenced samples and the number of good-quality sequences obtained. This difference may be due to various factors, such as a lower sensitivity of primers for different parts of the genome or the fact that the samples are from wild boar instead of tissue culture isolates. The highest discrepancy was observed for the IGR region, which represents a highly variable part of the ASFV genome, and the possibility of mutations at the primer binding site cannot be ruled out.
The I73R-I329L gene, together with the CVR, is one of the most variable TRS regions within the conserved central region of the ASFV genome (Bao et al. 2022), within which a “TATATAGGAA” repeat segment can be found (Gallardo et al. 2014). Through the analysis of the I73R-I329L gene, ASFV genotype II can be divided into four variants IGR I–IV, with variant I having two repeat segments, and variants II, III, and IV each having one more repeat segment than the previous one. According to the results described in this study, two different strains simultaneously circulating in the country have been found. In our study, the variant of ASFV's I73R-I329L gene with two TATATAGGAA repeat segments (referred to as the IGR II variant) was the most frequently detected (98.6%), which is in accordance with the majority of other genotype II strains (Gallardo et al. 2014, 2023; Tran et al. 2021; Nguyen et al. 2022; Shi et al. 2022). A novel IGR III variant has been detected in the country and additional samples were sequenced from the same hunting ground, as well as domestic pigs from backyard farms in the infected area, although additional IGR III variants have not been found. The IGR III variant has also been recorded in Poland (Mazur-Panasiuk et al. 2020) in an outbreak of domestic pigs, and in a case of a found dead wild boar, as well as in China (Shi et al. 2022), Vietnam (Tran et al. 2021; Nguyen et al. 2022) and South Korea (Kim et al. 2021). In all countries that have reported multiple IGR variants, the dominant variant remains the IGR II variant. The detected SNP mutation in the IGR is unique to the Serbian strains and could allow for a further differentiation of IGR II variants in which it has been detected.
The novel IGR III strain of the virus was detected in a pig herd in a region where no previous cases of the virus had been reported. Despite efforts to trace the source of the virus, no links were found to infected pigs in the area, making it difficult to determine how the virus was introduced. However, it is possible that the strain arose as a result of a single isolated mutation, although this is considered to be unlikely. Given the lack of effective measures to prevent the spread of ASFV strains from other affected districts, it is possible that humans could introduce novel strains of the virus into the area. This could occur through the transport of infected animals or contaminated animal products, highlighting the need for improved biosecurity measures to stop the introduction of novel strains of the virus. The exact evolutionary mechanism of ASFV remains unknown, and as such further studies are necessary in order to better understand the virulence, and the genomic characteristics of the virus. Human activities including illegal transport of infected animals or contaminated products across borders could be responsible for the emergence of new ASFV strains in new areas. Reports have linked the spread of ASFV by humans across borders or from infected zones in one part of the country to ASF-free regions with previous outbreaks in domestic pigs not connected to wild boars. It is speculated that the first case of ASFV in Serbia was linked to illegal transportation across the border (Milićević et al. 2019). This underscores the importance of enforcing stricter regulations and border controls to prevent the spread of the disease.
Conclusion
This study aimed to investigate the circulating strains of ASFV in Serbia by analyzing the CVR region and the IGR between the I73R and I329L genes. To achieve this, we conducted a comprehensive analysis of several different strains of ASFV circulating in the country. Since the initial introduction of ASFV into Serbia, all strains of the virus that have been identified in the country belong to genotype II and CVR I group, and the IGR II group. The findings of this study suggest that there is a co-circulation of at least three different CVR I strains, and concerning the IGR, this is a first report of a novel IGR III group detected within the country. These findings contributed to the knowledge of circulating strains of ASFV in Europe. Further research on other variable regions of the ASFV genome is necessary to better understand ASFV spread and evolution. Although whole genome sequencing (WGS) provides a high quantity of valuable information, it remains difficult and laborious, and the amount of quality information obtained concerning ASFV is limited. The size of the ASFV genome and the possible changes in the sense of an SNP or the repeat regions of a single strain make WGS challenging to apply successfully in the context of ASFV. Therefore, analyzing small, highly variable genomic markers remains the best cost-to-acquisition ratio for studying ASFV.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Achenbach JE, Gallardo C, Nieto-Pelegrín E et al (2017) Identification of a New Genotype of African Swine Fever Virus in Domestic Pigs from Ethiopia. Transbound Emerg Dis 64:1393–1404. https://doi.org/10.1111/tbed.12511
Alonso C, Borca M, Dixon L et al (2018) ICTV Virus Taxonomy Profile: Asfarviridae. J Gen Virol 99:613–614. https://doi.org/10.1099/jgv.0.001049
Bao J, Zhang Y, Shi C et al (2022) Genome-Wide Diversity Analysis of African Swine Fever Virus Based on a Curated Dataset. Animals 12:2446. https://doi.org/10.3390/ani12182446
Bastos ADS, Penrith M-L, Crucière et al (2003) Genotyping field strains of African swine fever virus by partial p72 gene characterisation. Arch Virol 148:693–706
Biek R, Pybus OG, Lloyd-Smith JO, Didelot X (2015) Measurably evolving pathogens in the genomic era. Trends Ecol Evol 30:306–313. https://doi.org/10.1016/j.tree.2015.03.009
Boshoff CI, Bastos ADS, Gerber LJ, Vosloo W (2007) Genetic characterisation of African swine fever viruses from outbreaks in southern Africa (1973–1999). Vet Microbiol 121:45–55. https://doi.org/10.1016/j.vetmic.2006.11.007
Cadenas-Fernández E, Ito S, Aguilar-Vega C et al (2022) The Role of the Wild Boar Spreading African Swine Fever Virus in Asia: Another Underestimated Problem. Front Vet Sci 9:453. https://doi.org/10.3389/FVETS.2022.844209/BIBTEX
Chenais E, Depner K, Guberti V et al (2019) Epidemiological considerations on African swine fever in Europe 2014–2018. Porcine Health Manag 5:6. https://doi.org/10.1186/s40813-018-0109-2
de la Vega I, González A, Blasco R et al (1994) Nucleotide Sequence and Variability of the Inverted Terminal Repetitions of African Swine Fever Virus DNA. Virology 201:152–156. https://doi.org/10.1006/viro.1994.1277
de Oliveira RP, Hutet E, Paboeuf F, et al (2019) Comparative vector competence of the Afrotropical soft tick Ornithodoros moubata and Palearctic species, O. erraticus and O. verrucosus, for African swine fever virus strains circulating in Eurasia. PLoS One 14:e0225657 https://doi.org/10.1371/JOURNAL.PONE.0225657
Denstedt E, Porco A, Hwang J et al (2021) Detection of African swine fever virus in free-ranging wild boar in Southeast Asia. Transbound Emerg Dis 68:2669–2675. https://doi.org/10.1111/TBED.13964
Department for Environment F and RAA and PHAAH and WA-IDM (2023) Updated Outbreak Assessment #24 - African swine fever (ASF) in Asia and Oceania. London
Dixon LK, Chapman DAG, Netherton CL, Upton C (2013) African swine fever virus replication and genomics. Virus Res 173:3–14. https://doi.org/10.1016/j.virusres.2012.10.020
European Centre for Disease Prevention and Control (ECDC) (2018) Ornithodoros spp. - current known distribution, May 2018
European Commission: Animal Disease Information System (2019) ADIS 2019
European Commission: Animal Disease Information System (2020) ADIS 2020
European Commission: Animal Disease Information System (2021) ADIS 2021
European Commission: Animal Disease Information System (2022) ADIS 2022
Faburay B (2022) Genome Plasticity of African Swine Fever Virus: Implications for Diagnostics and Live-Attenuated Vaccines. Pathogens 11:145. https://doi.org/10.3390/pathogens11020145
Forth JH, Calvelage S, Fischer M et al (2023) African swine fever virus – variants on the rise. Emerg Microbes Infect 12:2146537. https://doi.org/10.1080/22221751.2022.2146537
Forth JH, Forth LF, Blome S, et al (2020) African swine fever whole-genome sequencing—Quantity wanted but quality needed. PLoS Pathog 16:e1008779 https://doi.org/10.1371/journal.ppat.1008779
Gallardo C, Casado N, Soler A et al (2023) A multi gene-approach genotyping method identifies 24 genetic clusters within the genotype II-European African swine fever viruses circulating from 2007 to 2022. Front Vet Sci 10:1112850. https://doi.org/10.3389/fvets.2023.1112850
Gallardo C, Fernández-Pinero J, Pelayo V et al (2014) Genetic Variation among African Swine Fever Genotype II Viruses, Eastern and Central Europe. Emerg Infect Dis 20:1544–1547. https://doi.org/10.3201/eid2009.140554
Gallardo C, Mwaengo DM, Macharia JM et al (2009) Enhanced discrimination of African swine fever virus isolates through nucleotide sequencing of the p54, p72, and pB602L (CVR) genes. Virus Genes 38:85–95. https://doi.org/10.1007/s11262-008-0293-2
Gallardo C, Nurmoja I, Soler A et al (2018) Evolution in Europe of African swine fever genotype II viruses from highly to moderately virulent. Vet Microbiol 219:70–79. https://doi.org/10.1016/J.VETMIC.2018.04.001
Glišić D, Milićević V, Veljović L et al (2023) Patterns of ASFV Transmission in Domestic Pigs in Serbia. Pathogens 12:149. https://doi.org/10.3390/pathogens12010149
Gogin A, Gerasimov V, Malogolovkin A, Kolbasov D (2013) African swine fever in the North Caucasus region and the Russian Federation in years 2007–2012. Virus Res 173:198–203. https://doi.org/10.1016/j.virusres.2012.12.007
Iscaro C, Dondo A, Ruocco L et al (2022) Index case of new African Swine Fever incursion in mainland Italy. Transbound Emerg Dis. https://doi.org/10.1111/tbed.14584
Jean-Pierre RP, Hagerman AD, Rich KM (2022) An analysis of African Swine Fever consequences on rural economies and smallholder swine producers in Haiti. Front Vet Sci 9:1293. https://doi.org/10.3389/FVETS.2022.960344/BIBTEX
Jo YS, Gortázar C (2021) African Swine Fever in wild boar: Assessing interventions in South Korea. Transbound Emerg Dis 68:2878–2889. https://doi.org/10.1111/TBED.14106
Kim S, Lee S, Jeong H et al (2021) Rapid emergence of African swine fever virus variants with different numbers of a tandem repeat sequence in South Korea. Transbound Emerg Dis 68:1726–1730. https://doi.org/10.1111/tbed.13867
King DP, Reid SM, Hutchings GH et al (2003) Development of a TaqMan® PCR assay with internal amplification control for the detection of African swine fever virus. J Virol Methods 107:53–61. https://doi.org/10.1016/S0166-0934(02)00189-1
Koeltz A, Kolbasov D, Titov I, et al (2018) African Swine Fever Virus, Siberia, Russia, 2017. Emerg Infect Dis • www.cdc.gov/eid • 24 https://doi.org/10.1016/j.idcr.2014.12.002
Ladoși I, Păpuc TA, Ladoși D (2023) The impact of african swine fever (ASF) on Romanian pig meat production: A Review. Acta Vet-Bel 73:1–12. https://doi.org/10.2478/acve-2023-0001
Li X, Xiao K, Zhang Z et al (2020) The recombination hot spots and genetic diversity of the genomes of African swine fever viruses. J Infect 80:121–142. https://doi.org/10.1016/j.jinf.2019.08.007
Mazloum A, van Schalkwyk A, Chernyshev R et al (2023a) A Guide to Molecular Characterization of Genotype II African Swine Fever Virus: Essential and Alternative Genome Markers. Microorganisms 11:642. https://doi.org/10.3390/MICROORGANISMS11030642/S1
Mazloum A, Van Schalkwyk A, Chernyshev R et al (2022) Genetic Characterization of the Central Variable Region in African Swine Fever Virus Isolates in the Russian Federation from 2013 to 2017. Pathogens 11:919. https://doi.org/10.3390/pathogens11080919
Mazloum A, van Schalkwyk A, Shotin A et al (2023b) Whole-genome sequencing of African swine fever virus from wild boars in the Kaliningrad region reveals unique and distinguishing genomic mutations. Front Vet Sci 9:1976. https://doi.org/10.3389/FVETS.2022.1019808/BIBTEX
Mazur-Panasiuk N, Walczak M, Juszkiewicz M, Woźniakowski G (2020) The Spillover of African Swine Fever in Western Poland Revealed Its Estimated Origin on the Basis of O174L, K145R, MGF 505–5R and IGR I73R/I329L Genomic Sequences. Viruses 12:1094. https://doi.org/10.3390/v12101094
Mazur-Panasiuk N, Woźniakowski G, Niemczuk K (2019) The first complete genomic sequences of African swine fever virus isolated in Poland. Sci Rep 9:4556. https://doi.org/10.1038/s41598-018-36823-0
Milićević V, Kureljušić B, Maksimović Zorić J et al (2019) First Occurence of African Swine Fever in Serbia. Acta Vet Brno 69:443–449. https://doi.org/10.2478/acve-2019-0038
Nešković M, Ristić B, Došenović R et al (2021) African Swine Fever Outbreak Investigation on Large Commercial Pig Farm in Serbia. Acta Vet Brno 71:219–229. https://doi.org/10.2478/acve-2021-0019
Nguyen VT, Cho K, Mai NTA et al (2022) Multiple variants of African swine fever virus circulating in Vietnam. Arch Virol 167:1137–1140. https://doi.org/10.1007/s00705-022-05363-4
Nix RJ, Gallardo C, Hutchings G et al (2006) Molecular epidemiology of African swine fever virus studied by analysis of four variable genome regions. Arch Virol 151:2475–2494. https://doi.org/10.1007/s00705-006-0794-z
Nurmoja I, Petrov A, Breidenstein C et al (2017) Biological characterization of African swine fever virus genotype II strains from North-Eastern Estonia in European wild boar. Transbound Emerg Dis 64:2034–2041. https://doi.org/10.1111/TBED.12614
Oļševskis E, Guberti V, Seržants M et al (2016) African swine fever virus introduction into the EU in 2014: Experience of Latvia. Res Vet Sci 105:28–30. https://doi.org/10.1016/J.RVSC.2016.01.006
Pereira De Oliveira R, Hutet E, Lancelot R et al (2020) Differential vector competence of Ornithodoros soft ticks for African swine fever virus: What if it involves more than just crossing organic barriers in ticks? Parasit Vectors 13:1–15. https://doi.org/10.1186/S13071-020-04497-1/FIGURES/6
Pikalo J, Schoder M, Sehl J et al (2020) The African swine fever virus isolate Belgium 2018/1 shows high virulence in European wild boar. Transbound Emerg Dis 67:1654–1659. https://doi.org/10.1111/tbed.13503
Polaček V, Mirčeta J, Prodanov-Radulović J (2021) Key risk factors and impact of African swine fever spreading on pig production in Serbia. Acta Vet Bel 71:371–391. https://doi.org/10.2478/acve-2021-0032
Prodanov-Radulović J, Mirčeta J, Djurdjević B, et al (2023) African Swine Fever Outbreak in an Enclosed Wild Boar Hunting Ground in Serbia. Pathogens 12:691https://doi.org/10.3390/PATHOGENS12050691
Quembo CJ, Jori F, Vosloo W, Heath L (2017) Genetic characterization of African swine fever virus isolates from soft ticks at the wildlife/domestic interface in Mozambique and identification of a novel genotype. https://doi.org/10.1111/tbed.12700
Quembo CJ, Jori F, Heath L et al (2016) Investigation into the Epidemiology of African Swine Fever Virus at the Wildlife - Domestic Interface of the Gorongosa National Park, Central Mozambique. Transbound Emerg Dis 63:443–451. https://doi.org/10.1111/tbed.12289
Ramishvili Levan (2007) African swine fever - Georgia: OIE. In: ProMED INTERNATIONAL SOCIETY FOR INFECTIOUS DISEASES. https://promedmail.org/promed-post/?id=12219. Accessed 27 Mar 2023
Rowlands RJ, Michaud V, Heath L et al (2008) African Swine Fever Virus Isolate, Georgia, 2007. Emerg Infect Dis 14:1870–1874. https://doi.org/10.3201/eid1412.080591
Sargsyan MA, Voskanyan HE, Karalova EM, et al (2018) Third wave of African swine fever infection in Armenia: Virus demonstrates the reduction of pathogenicity. Vet World https://doi.org/10.14202/vetworld.2018.5-9
Sauter-Louis C, Conraths FJ, Probst C et al (2021a) African Swine Fever in Wild Boar in Europe—A Review. Viruses 13:1717. https://doi.org/10.3390/v13091717
Sauter-Louis C, Forth JH, Probst C et al (2021b) Joining the club: First detection of African swine fever in wild boar in Germany. Transbound Emerg Dis 68:1744–1752. https://doi.org/10.1111/TBED.13890
Shi K, Liu H, Yin Y, et al (2022) Molecular Characterization of African Swine Fever Virus From 2019–2020 Outbreaks in Guangxi Province, Southern China. Front Vet Sci 9:912224 https://doi.org/10.3389/fvets.2022.912224
Sun E, Huang L, Zhang X et al (2021) Genotype I African swine fever viruses emerged in domestic pigs in China and caused chronic infection. Emerg Microbes Infect 10:2183–2193. https://doi.org/10.1080/22221751.2021.1999779
Tran HTT, Truong AD, Dang AK et al (2021) Circulation of two different variants of intergenic region (IGR) located between the I73R and I329L genes of African swine fever virus strains in Vietnam. Transbound Emerg Dis 68:2693–2695. https://doi.org/10.1111/tbed.13996
Vilem A, Nurmoja I, Niine T et al (2020) Molecular Characterization of African Swine Fever Virus Isolates in Estonia in 2014–2019. Pathogens 9:582. https://doi.org/10.3390/pathogens9070582
WOAH (2022) WAHIS. https://wahis.woah.org/#/in-event/4761/dashboard. Accessed 27 Mar 2023
Woźniakowski G, Pejsak Z, Jabłoński A (2021) Emergence of African Swine Fever in Poland (2014–2021). Successes and Failures in Disease Eradication. Agriculture 11:738https://doi.org/10.3390/AGRICULTURE11080738
Acknowledgements
The sequences were generated through the Sequencing Service of the Animal Production and Health Sub-Programme of the Joint FAO/IAEA Division in Vienna, Austria.
Funding
The Serbian Ministry of Science, Technological Development and Innovation provided funding for the study through Contract No 451–03-47/2023–01/200030 and 451–03-47/2023–01/200143.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. D.G. was the principal investigator, molecular tests were carried out by D.G. and V.M., sequences were analysed by I.T. and C.G., the epidemiological investigation was carried out by R.P. and S.R., and D.K. and S.R. supervised the study.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
The published version of the manuscript has been read and agreed upon by all authors.
Competing interests
The authors state that they have no competing interests. The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Glišić, D., Milićević, V., Krnjaić, D. et al. Genetic analysis reveals multiple intergenic region and central variable region in the African swine fever virus variants circulating in Serbia. Vet Res Commun 47, 1925–1936 (2023). https://doi.org/10.1007/s11259-023-10145-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11259-023-10145-7