
Abstract
In unstructured dairy programs, pedigree is usually shallow, which leads to biased prediction of breeding values using best linear unbiased prediction (BLUP). The objective of this study was to come out with a genomic prediction strategy that can utilize shallow pedigree information and predict unbiased and more accurate GEBV for sex-limited traits in a small population using single-step GBLUP (ssGBLUP). The data and models for a population under selection were simulated. Out of current 10 generations, 10th generation with 1000 candidates served as validation population. For the complete pedigree scenario, pedigree (P)BLUP estimated breeding values (EBV) were unbiased with accuracy (r) of 0.35 ± 0.02 and 0.26 ± 0.01 for 0.3 and 0.1 h2 scenario, respectively. For the shallow pedigree, biased prediction of breeding values and low accuracies were obtained with linear decline in the accuracy of EBV for removal of information on more distant pedigree. Accuracy and bias (ρ) for scenario with removing 4 distant generations from pedigree were 0.30 ± 0.02 and 0.55 ± 0.03, respectively, in moderate h2 scenario. Use of Genomic (G)BLUP, especially with “extreme phenotypic contrast selective genotyping,” (TB) resulted in higher accuracy for a small reference of females; however, GEBV were highly biased. We observed that ssGBLUPF, where the numerator relationship matrix is corrected for inbreeding, resulted in more accurate and unbiased estimates of GEBV across shallow pedigree scenario, with TB all female reference (missing 4 distant generations: r = 0.50 ± 0.02; ρ = 0.96 ± 0.02). We recommend use of ssGBLUPF with two tailed selectively genotyped all female reference in shallow pedigree scenarios, to obtain unbiased and accurate GEBV for sex-limited traits, when resources are limited.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Genomic selection (GS) in the dairy industry has been accepted and replaced the traditional selection approach in most of the developed countries. The best linear unbiased prediction (BLUP) provides unbiased estimates of breeding values, even in populations under selection. However, inclusion of all information on the selection decisions is a necessary condition for obtaining such estimates (Henderson, 1975; Sorensen and Kennedy, 1984). In the absence of complete pedigree information, the predictions obtained from the pedigree-based BLUP (PBLUP) are biased. Usually, in the loose dairy breeding structure, the pedigree is not complete or present with holes. Research on implementation of genomic selection has shown that the single-step genomic best linear unbiased prediction (ssGBLUP) approach (Christensen & Lund 2010; Legarra et al. 2009) which combines the genomic relationship from genotyped animals having a pedigree with other non-genotyped animals in the pedigree results in use of genomic information and obtains better accuracies of prediction with unbiased breeding values (Gowane et al. 2019a). However, whether ssGBLUP can be useful in shallow pedigree situations with rather small population size needs to be explored.
The Indian dairy industry has a loose structure. Indigenous cattle have low production (3.41 kg/cow/day), as compared to crossbred cattle (7.33 kg/cow/day) (DAHDF, 2018). Low averages persist mostly due to low selection intensity in traditional breed improvement schemes and limited efforts for within breed selection to improve the indigenous population (Gowane et al. 2019b). Thus, the genetic improvement in these animals is highly desired. Cost cutting on genotyping for creation of a reference population is also an important criterion, when resources are less. In such scenarios, especially for dairy cattle breeding scenario in small populations, all female reference with selective genotyping of candidates from the current few generations is usually done. However, selection of these candidates affects the accuracy as well as bias of prediction. Selective genotyping of top and bottom phenotypically contrasting candidates have shown to improve accuracy of finding causative SNPs (van Gestel et al., 2000; Xu &Vogl, 2000; Huang & Lin, 2007). Cesarani et al. (2019) revealed impact of selective genotyping on the estimates of heritability and observed that estimates from GREML were more biased compared to those estimated by single-step (ss)GREML, because ssGREML was less affected by selective or limited genotyping. Gowane et al. (2019a) also observed that selective genotyping affect accuracy as well as bias of GEBV adversely using GBLUP; however, ssGBLUP results in better estimates. However, the question of how the selectively genotyped reference will have impact on ssGBLUP application on shallow pedigreed data is unclear. We are interested to quantify the effect of selective genotyping on GEBV in shallow pedigree scenarios.
This work was conceived to address above discussed issues. Here, we are trying to see utility of the ssGBLUP approach for implementation of GS especially when we have shallow pedigree information in small population scenario. Objectives of the study are (1) to observe the impact of shallow pedigree on the prediction of breeding values using pedigree BLUP versus GBLUP and ssGBLUP and (2) to study the effect of selective genotyping on ssGBLUP-derived GEBV in small population having shallow pedigree information.
Materials and methods
Simulation of data: population and genotype
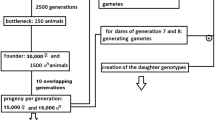
The QMSim simulator (Sargolzaei and Schenkel, 2009) was used for the data simulation. We have simulated a typical dairy cattle breeding scenario, with a sex-limited trait. Simulation involved 2 steps: (1) historical generations and (2) current generations. The historical population with 1000 generations was simulated with breeding structure, where 400 males mated 2000 females in generation 1 that reduced to 100 males and 1000 females in the last historical generation. Sex ratio was 1:1 for progeny born. In the current 10 generations (1001–1010), the breeding structure was 50 males mating with 1000 females producing 1000 progenies in each generation with 1:1 sex ratio. Replacement ratio for sires was 40%, and for dams, it was 20%. Top sires were selected in each current generation using estimated breeding values (EBV) as obtained by PBLUP. Top selected sires were mated to all the females randomly in each generation post selection. Selection was with non-overlapping generations. The scheme of simulation has been explained pictorially at Fig. 1.

Schematic representation of the simulation and the analytical methods used in this study
We simulated the genomic structure to match a 3 Giga base bovine genome, which were 30 chromosomes, each of 1 Morgan length. Across the genome, biallelic markers (N = 60,000) were distributed randomly with equal frequency of 0.5 in 1st generation of the historical population. According to Hickey and Gorjanc (2012), the mutation rate of the markers and QTL was fixed at 2.5 × 10−8 per locus per generation, and the number of crossovers was sampled from a Poisson distribution with positions randomly distributed. The simulated genetic model had 750 QTL for the trait and the QTL allele effects were sampled from a gamma distribution with shape parameter = 0.4 and scale = 1.0. The true breeding values (TBV) were obtained by aggregating genotype effects at individual QTL, which were re-scaled to match the input for the additive component of genetic variance.
These breeding values for candidates in the 1001st generation had a normal distribution with mean 0 and variance equal to heritability. For the phenotypes, the mean and variance were zero and one, respectively. 60 K biallelic marker data and phenotypes were simulated for all the animals (N = 11,050) in 10 generations, as we used only the 10th generation candidates for validation. The simulation study included two scenarios in terms of heritability (low = 0.10 and moderate = 0.30). For each animal from generation 1001–1010, pedigree, true breeding values, phenotypes, and genotypes were simulated, and breeding values were estimated. All the simulation scenarios were replicated 10 times, and the strategy and size of the training set were considered to be sufficient for the present aim of the study.
Breeding value prediction models
Pedigree-based prediction of breeding values
The pedigree-based BLUP (PBLUP) evaluations were made using following shallow pedigree scenarios. There were 10 generations of pedigree information, and the 10th generation was used for validation, where 1000 selection candidates were available which were used to determine the bias and accuracy of estimated breeding values.
Several models to match the shallow pedigree scenario were created for prediction of breeding values in 10th generation (n = 1000).
-
1.
Ped_10: involved using all the 10 generations (n = 10550 animals) pedigree information
-
2.
Ped_9: involved using current 9 generation (9550 animals) pedigree information
-
3.
Ped_8: involved using current 8 generation (8550 animals) pedigree information
-
4.
Ped_7: involved using current 7 generation (7550) pedigree information
-
5.
Ped_6: involved using current 6 generation (6550 animals) pedigree information
-
6.
Ped_5: involved using current 5 generation (5550 animals) pedigree information
-
7.
Ped_4: involved using current 4 generation (4550 animals) pedigree information
The linear mixed model was used for prediction of breeding value using PBLUP as given by Henderson (1975). As the trait for the analysis is milk yield, sex effect was not considered in the analysis. PBLUP involved animal model with direct animal additive effect as a random factor (a). The distributional assumptions of the model were a ~ N(0,Aσ2a) and e ~ N(0,Iσ2e); where A is numerator relationship matrix, I is an identity matrix, and σ2a and σ2e are additive genetic and residual variances, respectively.
The prediction accuracy (r) for breeding values was estimated by obtaining the Pearson correlation between the TBV and EBV of the validation dataset in 10th generation. The regression coefficient between the TBV and EBV indicated the over or under-dispersion of EBVs or bias of prediction (ρ).
Genomic information-based prediction models
Selective genotyping strategies
For the genomic selection strategy, the selective genotyping scenarios were created for creation of reference using only female information.
-
a.
Highest EBVs (T1000): From each of the last 4 generations (6 to 9), top ranking 250 females based on their EBV were selected for genotyping to form a reference population of 1000 animals (T1000).
-
b.
Two-tailed EBVs (TB1000): The top and bottom selective genotyping of phenotypically extreme/contrasting females constituted this subset. It represented a selection of 125 top and 125 bottom females based on their estimated breeding values in each of generation 6 to 9 to form a reference population of 1000 animals (TB1000).
The GBLUP analysis was used to estimate the genomic estimates of breeding values (GEBV). We subjected the genotype data for quality control (QC) by PLINK 1.9 (Chang et al. 2015). The QC resulted in exclusion of SNP with minor allele frequency < 0.05, and p value for Hardy–Weinberg equilibrium < 0.001. The genomic relationship matrix (G) was constructed using the software package MTG2 (Lee and van der Werf 2016), where we have used the option to obtain G according to Van Raden’s method 1. We could obtain the GEBV for 1000 candidates in the current generation 10 with MTG2 using information on their genotypes. The model used for genomic analysis GBLUP included the vector of additive genetic effects of animal (g), where g ~ N(0, Gσ2g). The fixed effects were not simulated except sex, which was not included in the analysis owing to the sex-limited nature of the trait.
Single-step GBLUP evaluation for shallow pedigree scenarios
To see the impact of extending the genotypic information to non-genotyped animals in the pedigree and its utility in shallow pedigree problem, we created several scenarios with combinations of shallow pedigree models and selectively genotyped references (T1000 or TB1000). Shallow pedigree models were where information on preceding pedigree was missing (Table 1).
Single-step genomic BLUP (ssGBLUP) combined the information from pedigree and genotypes where information was extended to non-genotyped relatives from the genotyped reference. We assumed that the variance components in the simulated data as known while using ssGBLUP method. The model for the ssGBLUP evaluation used the vector of direct additive genetic effects (u), which is assumed as normally distributed with u ~ N(0,Hσ2u). The H notifies a matrix which include both genotyped and non-genotyped individuals (Christensen and Lund 2010; Aguilar et al. 2010). The fixed effects except sex were not simulated, and hence fixed effect was not used owing to sex-limited nature of the trait.
For better compatibility between A and G in ssGBLUP, the modified A22 + (G*−1 − A22−1) with G* being αG + (1-α)A22 were used in the H−1 matrix. The value of α was 0.99. We also scaled G based on A22 by equating mean of diagonal of A22 with mean of diagonal of G. The method presented here did not include inbreeding in the A−1; however, inbreeding in A22−1 was considered.
Another single-step method (ssGBLUPF) was considered for analysis, where the A−1 was constructed by taking in to account the inbreeding of pedigree correctly (Garcia-Baccino et al. 2017). The inbreeding is used to create the variance of Mendelian sampling needed to create A−1. Without inbreeding in A−1, the compatibility among A−1, G−1, and A22−1 is compromised. Metafounder approach (Legarra et al. 2015) was of no value in this data owing to absence of incomplete ancestral relationship. We used the BLUPF90 programs (Misztal 2008; Aguilar et al. 2014) for all the single-step models.
The prediction accuracy for breeding values was estimated by obtaining the Pearson correlation between the TBV and (G)EBV of the validation dataset in 10th generation. The regression coefficient between the TBV and (G)EBV indicated the over or under-dispersion of (G)EBVs or bias of prediction.
Results
Simulated population
We have simulated two scenarios (heritability = 0.10 or 0.30). High LD estimates were observed for SNPs located in close proximity. The mean r2 for pairs of SNP closer than 0.1 Mb was 0.31 for generation 1 and 0.34 and 0.35 for generation 10 in low and moderate h2 scenarios, respectively. Similarly, for distance between 0.1 and 1 Mb, the average r2 was 0.19. Disequilibrium decreased sharply with increasing distance between SNP. All chromosomes were simulated using the same parameters, and therefore differences between them were not expected. In the medium and low-heritability scenarios, the average inbreeding coefficients in the last generation were 0.05 ± 0.003 and 0.07 ± 0.004, respectively, and the average accuracies of the pedigree indices were 0.51 ± 0.01 and 0.35 ± 0.02, respectively. As the selection of candidates in current generations was on the basis of EBV, the increase in the phenotypes of females over the generations was observed. For the h2 = 0.1 scenario, average phenotype improved from 0.014 ± 0.008 in generation 1 to 0.739 ± 0.019 in generation 10. For the h2 = 0.3 scenario, the average phenotype improved from − 0.002 ± 0.007 in generation 1 to 1.80 ± 0.05 in generation 10.
Impact of shallow pedigree on the prediction of breeding values using PBLUP
BLUP uses additive genetic variance and residual variance information which actually scales the estimates of breeding values. Complete pedigree in our data had two scenarios (0.1 and 0.3 h2). However, we have seen that the shallow pedigrees affect the estimate of additive genetic variance (Va) and residual variance (Ve) significantly. Table 2 shows that the with every one generation information missing, the estimates were unpredictable and did not follow a particular trend. However, they were no more representing the actual whole pedigree genetic variance, but only the variance for the data available. This has also affected the estimates of breeding values. Fluctuations were much higher for 0.3 h2 scenario as compared to 0.1 h2 scenario.
Prediction of breeding values for the validation population (10th generation) with N = 1000, using PBLUP revealed that the accuracy of prediction was 0.35 ± 0.02 and the bias of prediction was 0.98 ± 0.05, when information from all the pedigrees (Ped_10) was utilized for the analysis in scenario of moderate heritability (h2 = 0.30). A compromised accuracy (0.26 ± 0.01) was observed for the low heritability scenario (h2 = 0.10); however, prediction bias was not observed. The results revealed that the prediction accuracy is optimum and also the bias was negligible as the pedigree could trace back to the base population (Table 3).
The same dataset was again analyzed with shallow pedigree scenarios. Results revealed that there was a linear decline in the accuracy of the prediction and increase in the prediction bias with removal of information on more distant pedigree relatives from the data (Table 3) for low and moderate heritability scenarios. We observed that with current 5 generations of data available in the pedigree (Ped_5) and removing preceding 5 generations, the accuracy fell to 0.30 ± 0.02, which is 14.29% decline in the accuracy as compared to Ped_10 in moderate heritability scenario. Similarly, for the bias of prediction, we observed the estimate of regression of TBV on EBV was 0.43 ± 0.03. This indicated that bias increased by 56.12% due to the shallow pedigree effect. For the low h2 scenario, the decline from Ped_10 to Ped_5 for accuracy was 23% and bias increased by 61.9%, with over-dispersed EBVs.
Impact of using only genomic information on accuracy and bias of GEBV
For GS, we created the reference population (N = 1000) by selectively genotyping the top-ranking females (T1000) from the last 4 generations versus top and bottom selective genotyping of females (TB1000). The estimates of additive genetic variance were affected significantly in the selectively genotyped reference populations. We observed that the T1000 had 0.14 ± 0.03, whereas TB1000 had 2.02 ± 0.05 Va for 0.3 h2 scenario. For 0.1 h2 scenario, T1000 and TB1000 had 0.08 ± 0.02 and 1.09 ± 0.02 as Va. Estimates of Va were inflated due to two-tailed selective genotyping across heritability scenarios (Table 2).
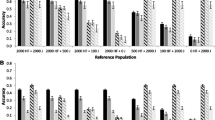
The results revealed that the exclusion of pedigree information and utilizing only genomic information on the reference animals could predict the breeding values with an accuracy of 0.27 ± 0.05 for T1000 (Fig. 1). The prediction bias was very high (1.79 ± 0.44) and under-dispersed the GEBV in moderate h2 scenario. For top and bottom selective genotyping, we observed significant improvement in the accuracy (0.50 ± 0.02); however, huge bias still existed (over-dispersion of GEBV). Improved accuracy using TB1000 was 85.18% over T1000 and 42.86% as compared to Ped_10 scenarios. For the low heritability scenario, we observed a similar trend; however, the estimates were low (Fig. 2 and Fig. 3). T1000 was highly biased, and accuracy was only 0.04; however, with TB1000, the accuracy rose to 0.34 ± 0.02, but again the estimates were highly biased.

Accuracy of breeding value prediction using different prediction methods and approaches. (Ped_10): The scenarios involved using all the 10 generations (n = 11,050 animals) pedigree for prediction of breeding values in 10th generation (n = 1000), (Ped_9): using current 9 generation (10,050 animals) pedigree, (Ped_8): using current 8 generation (9550 animals) pedigree, (Ped_7): using current 7 generation (8550 animals) pedigree, (Ped_6): using current 6 generation (7550 animals) pedigree, (Ped_5): using current 5 generation (6550 animals) pedigree. T1000: using 250 top performing females (selective genotyping) from each generation 6 to 9: n = 1000, in the reference for GBLUP. TB1000: using 125 top and 125 bottom performing females (selective genotyping) from each generation 6 to 9: n = 1000, in the reference for GBLUP. H10TB: Using 10 current generations pedigree and genomic relationships from TB1000 in the reference. H9TB: using 9 current generations pedigree assuming the previous generations are not known and genomic relationships from TB1000 in the reference. H8TB: using 8 current generations pedigree assuming the previous generations are not known and genomic relationships from TB1000 in the reference. H7TB: using 7 current generations pedigree assuming the previous generations are not known and genomic relationships from TB1000 in the reference. H6TB: using 6 current generations pedigree assuming the previous generations are not known and genomic relationships from TB1000 in the reference

Bias of breeding value prediction using different prediction methods and approaches. (Ped_10): The scenarios involved using all the 10 generations (n = 11,050 animals) pedigree for prediction of breeding values in 10th generation (n = 1000), (Ped_9): using current 9 generation (10,050 animals) pedigree, (Ped_8): using current 8 generation (9550 animals) pedigree, (Ped_7): using current 7 generation (8550 animals) pedigree, (Ped_6): using current 6 generation (7550 animals) pedigree, (Ped_5): using current 5 generation (6550 animals) pedigree. T1000: using 250 top performing females (selective genotyping) from each generation 6 to 9: n = 1000, in the reference for GBLUP. TB1000: using 125 top and 125 bottom performing females (selective genotyping) from each generation 6 to 9: n = 1000, in the reference for GBLUP. H10TB: using 10 current generations pedigree and genomic relationships from TB1000 in the reference. H9TB: using 9 current generations pedigree assuming the previous generations are not known and genomic relationships from TB1000 in the reference. H8TB: using 8 current generations pedigree assuming the previous generations are not known and genomic relationships from TB1000 in the reference. H7TB: using 7 current generations pedigree assuming the previous generations are not known and genomic relationships from TB1000 in the reference. H6TB: using 6 current generations pedigree assuming the previous generations are not known and genomic relationships from TB1000 in the reference
Single-step BLUP for shallow pedigree predicted unbiased GEBV
The single-step scenarios in our study involved combining all pedigree (10 current generations) with genotyped reference T1000 or TB1000. The holes in the pedigree scenario prevailed here too. We created the shallow pedigree scenario in ssGBLUP by removing one distant generation every scenario. The moderate heritability scenario (0.30) revealed significant improvement in accuracy of GEBV prediction as compared to GBLUP or PBLUP scenario. We observed that using inbreeding of A in single-step (ssGBLUPF) resulted in much better estimates for bias of GEBV. Therefore, we present results of only ssGBLUPF henceforth. In top selective genotyping scenario for ssGBLUPF, accuracy was higher (0.43) for H10T that reduced linearly for every distant generation pedigree removal from the dataset. For H6T, the accuracy was 0.35, which was 18.6% less than all pedigree scenario. However, even with shallow pedigree for 6–9 generations alone, the H6T could yield accuracy of 0.35, which was as good as the Ped_10 scenario. This reveals the predictive power of ssGBLUPF even with small genotyped reference. Not only this but also, the bias of prediction of GEBV was minimum with ssGBLUPF method across complete versus shallow pedigree scenarios. The predictions were unbiased for 2 to 3 distant generations missing pedigree (H10T, H9T, H8T); however, after that the larger the proportion of missing pedigree, the more over-dispersion of GEBV was observed. For H6T, nearly 8% biased estimates were obtained as compared to H9T or H10T (Table 4).
Significantly better estimates for top–bottom selective genotyping with ssGBLUPF approach were observed. High estimates of accuracy of GEBV (0.51) were observed for H10TB to H8TB. This was reduced to 0.50 for H7TB and H6TB. The estimate for H6TB was 16% higher than H10T, 26% higher than H6T, and 30% higher than the Ped_10 scenario. The accuracy from top–bottom ssGBLUPF was similar to the GBLUP estimates of TB1000; however, unlike TB1000, the estimates from ssGBLUPF were not biased. We observed regression of TBV on GEBV ranging from 0.96 to 1.00 across ssGBLUPF scenarios.
For low heritability scenarios, similar trend of estimates was observed. However, the estimates for accuracy using ssGBLUPF with TB1000 reference were higher than PBLUP complete pedigree model (Ped_10: r = 0.26) by 23.5% across the shallow pedigree scenarios (r = 0.34 for H6TB). Estimate for ρ was nearly 0.9 across ssBLUPF shallow pedigree models (Fig. 2 and Fig. 3).
Discussion
Linkage disequilibrium (LD) between adjacent markers is a criterion for quality control of the simulation before genomic evaluations are done. The level of LD decay with respect to the distance between SNPs, inbreeding levels in current generations along with estimates for the accuracy of traditional genetic evaluations, was also evaluated. High LD estimates were observed for SNPs located in close proximity. From the literature, we observed that the mean r2 estimate of 0.19 was pretty close to 0.18 in Sahiwal cattle (Mustafa et al. 2018), where, in congruence to this study, pairs of SNP closer than 0.1 Mb had similar r2 (0.35) which reduced with increase in distance between the SNP marker pairs. Not many studies on Indigenous cattle are available to compare the estimates; however, values of r2 between 0.20 and 0.31 have been reported for different cattle populations (Sargolzaei et al., 2008; Bohmanova et al. 2010; Banos and Coffey, 2010; Habier et al., 2010) as we reported in the simulated data.
The inbreeding and pedigree index accuracies were also within the range of estimates reported for dairy cattle populations for real data (Kearney et al., 2004; Gonzalez-Recio et al., 2006; Gonzalez-Recio et al., 2007, VanRaden et al., 2009a). In the medium and low-heritability scenarios, the average inbreeding coefficients in the last generation were 0.05 ± 0.003 and 0.07 ± 0.004, respectively, and the average accuracies of the pedigree indices were 0.51 ± 0.01 and 0.35 ± 0.02, respectively. As the selection of candidates in current generations was on the basis of EBV, the increase in the phenotypes of females over the generations was observed for the h2 = 0.1 scenario, and the average phenotype improved from 0.014 ± 0.008 in generation 1 to 0.739 ± 0.019 in generation 10. For the h2 = 0.3 scenario, the average phenotype improved from − 0.002 ± 0.007 in generation 1 to 1.80 ± 0.05 in generation 10.
Using ssGBLUPF approach for shallow pedigree information in small populations can result in more accurate and unbiased estimate of GEBV
BLUP accounts for culling, selection effects, and non-random mating provided the non-selected animals and mates are included in the analysis (Henderson 1975; Sorensen and Kennedy; 1984; Kennedy et al. 1988). Not including relationships to the unselected base population in the analysis will lead to selection bias (Van der Werf & De Boer, 1990). We observe that for the PBLUP analysis, the prediction accuracy is optimum, and also the bias was negligible as the pedigree could trace back to the base population, when all the pedigree was used for the prediction of breeding values. In our study, inconsistent estimates of Va and Ve for shallow pedigree scenarios across low and moderate heritability scenarios were observed, when complete pedigree information was not used for estimation purpose. Gowane et al. (2016) in real data on live weight and fleece weight of sheep have shown similar trend, as truncated data only represent the information hidden in it and do not trace back to the founder population. However, removing distant pedigree information resulted in loss of accuracy as well as increase in bias of EBV across low and medium h2 scenarios. Increase in bias of prediction occurs mostly due to absence of the selection and culling decision data from the pedigree and inability to trace it back to the base population. Clark et al. (2012) reported no significant difference in accuracy between shallow and deep pedigree BLUP estimated EBVs when the animals in the test and reference datasets had a close relationship; however, numerically the deep PBLUP yielded better results. Even for declining relationship between the test and reference datasets, deep pedigree BLUP proved better.
Use of only genomic information for prediction of GEBV resulted in more accurate GEBV estimates. The TB1000 scenario for GBLUP proved better than T1000 for prediction of GEBV, owing to increase in the power to detect the putative QTL for the trait of interest. Jiménez-Montero et al. (2012) also reported that the magnitude of prediction accuracy of GEBV depends on the number of animals genotyped and the selective genotyping strategy used. Literature also reveals that the worst prediction accuracies when genotypes only for the animals with the best phenotypes were included in the model (Jenko et al., 2017). Pryce et al. (2012) and Gao et al. (2015) suggested that the best strategy to include genotypes in genomic selection schemes is to choose random or unselected females. Two-tailed selective genotyping of candidates for phenotypic contrasts greatly improves the power to detect and map QTL in genetic association studies (Darvasi & Soller, 1992; van Gestel et al., 2000; Henshall & Goddard, 1999; Huang & Lin, 2007; Lebowitz et al., 1987; Muranty & Goffinet, 1997; Xu & Vogl, 2000), owing to which TB1000 proved better. Boligon et al. (2012) and Perez et al. (2018) reported that inclusion of extreme phenotypes in the reference is useful to accurately predict the most important SNPs in genome‐wide association studies, although with inflated GEBV. In small population, where the pedigrees are likely to be incomplete, the prediction accuracy was shown to be increased by including genotypes from cows with phenotypic records (Jenko et al., 2017; Pryce et al., 2012) using GBLUP.
The (co)variances for true breeding values for selected candidates are not well described by relationship matrix unless all records used in selection are accounted for. Thus GBLUP, although lead to increase in the accuracy but also resulted in biased GEBV. We also observed the biased estimates of Va and Ve in selectively genotyped reference. Higher and inflated estimates were observed for TB1000 as compared to T1000, which has also affected the accuracy of prediction and caused bias of prediction of breeding values. Cesarani et al. (2019) observed biased estimates of heritability for GREML as compared to SSGREML in selectively genotyped reference. BLUP required estimates for lambda (σ2e/σ2a) for prediction of breeding values. Biased estimates of variance components will affect the EBV magnitude. Several studies by early researchers have shown similar results, where using information only on the current generations for genotyping has resulted in significant bias of prediction (Van Raden et al. 2009a and 2009b; Patry and Ducrocq 2011; Vitezica et al. 2011; Gowane et al. 2019a) and use of single-step approach has been utilized as a strategy to address the issue of low accuracy and prediction bias (Gowane et al. 2019a). However, single-step strategy is beneficial when we have good pedigree that is deep enough so that the genomic relationship is extended to the non-genotyped animals readily. In case of the shallow pedigree issue in small populations, this option is explored in this study.
We observed improved accuracy as well as fairly unbiased estimates of GEBV when ssGBLUPF approach having TB1000 as reference was used in low as well as moderate h2 scenarios. Incomplete pedigree issues result in not accounting for complete ancestral relationship in NRM as it is given by the GRM. We observed that correction for pedigree inbreeding resulted in more accurate and unbiased estimates of GEBV. Similar observations were given by Garcia-Baccino et al. (2017) and Gowane et al. (2019a). Correction of NRM helps to bring the NRM and GRM at the same level for better merging. For shallow pedigree, ssGBLUPF proved to be the best strategy when TB1000 reference was genotyped. Use of extreme phenotypes in reference lead to better identification of marker effects, and hence the accuracy of prediction for GEBV was higher. Meyer (2021) showed that missing pedigrees significantly affected predicted breeding values in ssGBLUP and suggested unknown parent group (UPG) models with separation of genotyped and non-genotyped individuals and also the analyses fitting metafounders for better predictions of GEBV in terms of accuracy and bias.
Use of pedigree information, at least 1 extra generation than the genotyped reference, led to significant reduction in bias of prediction and accuracy as good as complete pedigree in ssGBLUPF in our study. Pocrnic et al. (2017) reported similar results, saying that including animals with phenotypic records in addition to 3 generations of pedigree was enough to obtain the same GEBV relative to using the full pedigree. We also observed that inclusion of more pedigree information in the single-step certainly improves the accuracy of prediction and also reduces the bias; however, the cost involved in recording deep pedigrees is a limiting factor. For the holes in pedigree situation, the accuracy obtained with ssGBLUPF seems optimal. Lourenco et al. (2014) in US Holsteins, Israeli Holsteins, and pig populations and Yang and Su (2016) in simulated data observed that in general, 3 generations of phenotypic data along with 2 ancestral generations in the pedigree were sufficient for prediction of the breeding values; however, it also depends on the trait h2 and population structure.
Conclusion
Phenotypic data maintained either in field or in farm in loose dairy breeding structure is usually not complete with respect to the pedigree information, and therefore, using complete pedigree information in genomic evaluation with ssGBLUPF is not possible. However, from our study, we conclude that even with last few generations selective genotyping (phenotypic contrasts) of 25% females to identify marker effects along with 2 generations of pedigree information will be necessary and sufficient to predict unbiased GEBV with optimum accuracy so as to match complete pedigree scenario in sex-limited traits. This strategy not only predicts better breeding values but also has potential to save huge costs on genotyping.
Data availability
The authors affirm that all data necessary for confirming the conclusions of the article are present within the article, figure, and tables. However, we have also attached the data in brief, where the parameter files for simulated data using QMSim are given. Any interested user can actually use these parameter files and replicate our results.
References
Aguilar, I., I. Misztal, Tsuruta, S., Legarra, A. & Wang, H. 2014. PREGSF90 – POSTGSF90: Computational Tools for the Implementation of Single-step Genomic Selection and Genome-wide Association with Ungenotyped Individuals in BLUPF90 Programs. Proceedings, 10th World Congress of Genetics Applied to Livestock Production.
Aguilar, I., Misztal, I., Johnson, D.-L., Legarra, A., Tsuruta, S., & Lawlor, T. J. 2010. Hot topic: A unified approach to utilize phenotypic, full pedigree, and genomic information for genetic evaluation of Holstein final score, Journal of Dairy Science, 93, 743-752. https://doi.org/10.3168/jds.2009-2730
Banos, G. &Coffey, M.P. 2010. Short communication: characterization of the genome-wide linkage disequilibrium in 2 divergent selection lines of dairy cows, Journal of Dairy Science, 93, 2775–2778. https://doi.org/10.3168/jds.2009-2613
Bohmanova, J., Sargolzaei M. & Schenkel F.S. 2010. Characteristics of linkage disequilibrium in North American Holsteins, BMC Genomics, 11: 421. https://doi.org/10.1186/1471-2164-11-421.
Boligon, A. A., Long, N., Albuquerque, L. G., Weigel, K. A., Gianola, D., & Rosa, G. J. M. 2012. Comparison of selective genotyping strategies for prediction of breeding values in a population under- going selection, Journal of Animal Science, 90, 4716–4722. https://doi.org/10.2527/jas.2012-4857
Cesarani, A., Pocrnic, I., Macciotta, N. P. P., Fragomeni, B. O., Misztal I., & Lourenco, D. A. L. 2019. Bias in heritability estimates from genomic restricted maximum likelihood methods under different genotyping strategies. Journal of Animal Breeding & Genetics, 136, 40–50. https://doi.org/10.1111/jbg.12367
Chang, C. C., Chow, C. C., Tellier, L. C. A. M., Vattikuti, S., Purcell, S. M. & Lee, J. J. 2015) Second-generation PLINK: rising to the challenge of larger and richer datasets, Giga Science, 4, https://www.cog-genomics.org/plink2.
Christensen, O. F. & Lund, M. S. 2010. Genomic prediction when some animals are not genotyped. Genetics Selection Evolution, 42:2, https://doi.org/10.1186/1297-9686-42-2.
Clark, S. A., Hickey, J. M., Daetwyler, H. D., & van der Werf, J. H. 2012. The importance of information on relatives for the prediction of genomic breeding values and the implications for the makeup of reference data sets in livestock breeding schemes, Genetics, selection, evolution, 44(1), 4. https://doi.org/10.1186/1297-9686-44-4
DAHDF. 2018. Annual report of department of animal husbandry, dairying and fisheries, ministry of agriculture, Government of India. Retrieved from ‘http://dadf.gov.in/sites/default/filess/annual_report_17-18.pdf’ on 22.05.2018
Darvasi, A., & Soller, M. 1992. Selective genotyping for determi- nation of linkage between a marker locus and a quantitative trait locus, Theoretical and Applied Genetics, 85, 353–359. https://doi.org/10.1007/BF00222881
Gao, H., Madsen, P., Nielsen, U.S., Aamand, G.P., Su, G., Byskov, K. & Jensen, J. 2015. Including different groups of genotyped females for genomic prediction in a Nordic Jersey population, Journal of Dairy Science, 98, 9051-9059. https://doi.org/10.3168/jds.2015-9947
Garcia-Baccino, C., A., Legarra, A., Christensen, O. F., Misztal, I., Pocrnic, I., Vitezica, Z. G. & Cantet, R. J. C. 2017. Metafounders are related to Fst fixation indices and reduce bias in single-step genomic evaluations. Genetics Selection Evolution, 49:34, https://doi.org/10.1186/s12711-017-0309-2
Gonzalez-Recio, O., Alenda, R., Chang, Y.M., Weigel, K. & Gianola, D. 2006.Selection for female fertility using censored fertility traits and investigation of the relationship with milk production. Journal of Dairy Science, 89, 4438–4444. https://doi.org/10.3168/jds.S0022-0302(06)72492-4
Gonzalez-Recio, O., Lopez de Maturana, E.& Gutierrez, J.P. 2007. Inbreeding depression on female fertility and calving ease in Spanish dairy cattle, Journal of Dairy Science, 90, 5744–5752. https://doi.org/10.3168/jds.2007-0203
Gowane, G. R., Lee, S. H., Clark, S., Moghaddar, N., Al-Mamun, H. A. & van der Werf, J. H. J. 2019a. Effect of selection and selective genotyping for creation of reference on bias and accuracy of genomic prediction. Journal of Animal Breeding and Genetics, 136, 390-407. https://doi.org/10.1111/jbg.12420
Gowane, G.R., Prince, L.L.L., Prakash, V., Sharma, R. C. & Kumar, A. 2016. Decline in Additive Genetic Variance for Live Weight and Greasy Fleece Weight in Bharat Merino Sheep, Agricultural research, 5, 316–323. https://doi.org/10.1007/s40003-016-0232-8
Gowane, G.R., Kumar, A. & Nimbkar, C. 2019b. Challenges and opportunities to livestock breeding programmes in India, Journal of Animal Breeding & Genetics, 136, 329-338. https://doi.org/10.1111/jbg.12391
Habier, D., Tetens, J., Seefried, F.R., Lichtner, P.& Thaller, G. 2010. The impact of genetic relationship information on genomic breeding values in German Holstein cattle, Genetics Selection Evolution, 42, 5. https://doi.org/10.1186/1297-9686-42-5
Henderson, C. R. 1975. Best linear unbiased estimation and prediction under a selection model. Biometrics, 31,423-447. https://doi.org/10.2307/2529430
Henshall, J. M., & Goddard, M. E. 1999. Multiple‐trait mapping of quantitative trait loci after selective genotyping using logistic regression, Genetics, 151, 885–894. https://doi.org/10.1093/genetics/151.2.885
Hickey, J. M. & Gorjanc, G. 2012. Simulated Data for Genomic Selection and Genome-Wide Association Studies Using a Combination of Coalescent and Gene Drop Methods. G3: Genes|Genomes|Genetics, 2, 425–427. https://doi.org/10.1534/g3.111.001297
Huang, B. E., & Lin, D. Y. 2007. Efficient association mapping of quantitative trait loci with selective genotyping, The American Journal of Human Genetics, 80, 567–576. https://doi.org/10.1086/512727
Jenko, J., Wiggans, G.R., Cooper, T.A., Eaglen, S.A.E., Luff, W.G.L., Bichard, M., Pong-Wong R. & Woolliams J.A. 2017. Cow genotyping strategies for genomic selection in a small dairy cattle population, Journal of Dairy Science 100, 439-452. https://doi.org/10.3168/jds.2016-11479
Kearney, J.F., Wall, E., Villanueva, B.& Coffey, M.P. 2004. Inbreeding trends andapplication of optimized selection in the UK Holstein population, Journal of Dairy Science, 87, 3503–3509.
Kennedy, B. W., Schaeffer, L. R., & Sorensen, D. A. 1988. Genetic properties of animal models, Journal of Dairy Science, 71, 17–26. https://doi.org/10.1016/S0022-0302(88)79975-0
Lebowitz, R. J., Soller, M., & Beckmann, J. S. 1987. Trait‐based analyses for the detection of linkage between marker loci and quantitative trait loci in crosses between inbred lines, Theoretical and Applied Genetics, 73, 556–562. https://doi.org/10.1007/BF00289194
Lee, S. H. & van der Werf, J. H. J. 2016. MTG2: An efficient algorithm for multivariate linear mixed model analysis based on genomic information, Bioinformatics, 32, 1420-1422. https://doi.org/10.1093/bioinformatics/btw012
Legarra, A., Aguilar, I. & Misztal, I. 2009. A relationship matrix including full pedigree and genomic information, Journal of Dairy Science, 92, 4656–4663. https://doi.org/10.3168/jds.2009-2061
Legarra, A., Christensen, O.F., Vitezica, Z.G., Aguilar, I. &Misztal, I. 2015. Ancestral relationships using metafounders: Finite ancestral populations and across population relationships, Genetics, 200, 455-468. https://doi.org/10.1534/genetics.115.177014
Lourenco, D. A. L., I. Misztal, S. Tsuruta, I. Aguilar, T. J. Lawlor, S. Forni, & J. I. Weller. 2014. Are evaluations on young genotyped animals benefiting from the past generations? Journal of Dairy Science, 97, 3930–3942. https://doi.org/10.3168/jds.2013-7769
Misztal, I. 2008. BLUPF90 - a flexible mixed model program in Fortran 90. Animal and Dairy Science, University of Georgia, August 2008.
Muranty, H., & Goffinet, B. 1997. Selective genotyping for location and estimation of the effect of a quantitative trait locus, Biometrics, 53, 629–643. https://doi.org/10.2307/2533963
Mustafa, H., Ahmad, N., Heather, H. J.,Eui-soo, K., Khan,W. A., Ajmal, A.,Javed, K., Pasha, T. N., Ali, A., Kim, J. J.& Sonstegard, T. S. 2018. Whole genome study of linkage disequilibrium in Sahiwal cattle, South African Journal of Animal Science, 48, 354-360. https://doi.org/10.4314/sajas.v48i2.16
Meyer, K. 2021. Impact of missing pedigrees in single-step genomic evaluation, Animal Production Science, https://doi.org/10.1071/AN21045
Patry, C. & Ducrocq, V. 2011. Evidence of biases in genetic evaluations due to genomic preselection in dairy cattle, Journal of Dairy Science, 94, 1011-1020. https://doi.org/10.3168/jds.2010-3804
Perez, B. C., Balieiro, J. C. C., Carvalheiro, R., Tirelo, F., Junior, G. A. O., Dementshuk, J. M., ... Ventura, R. V. 2018. Accounting for population structure in selective cow genotyping strategies, Journal of Animal Breeding and Genetics, 136, 23–39. https://doi.org/10.1111/jbg.12369
Pocrnic, I., Lourenco, D. A. L., Bradford, H., Chen, C. & Miztal, I. 2017. Technical note: Impact of pedigree depth on convergence of single-step genomic BLUP in a purebred swine population, Journal of Animal Science, 95, 3391-3395. https://doi.org/10.2527/jas.2017.1581
Pryce J. E., Hayes, B. J. & Goddard, M. E. 2012. Genotyping dairy females can improve the reliability of genomic selection for young bulls and heifers and provide farmers with new management tools. ICAR Conference, Cork, Ireland. Accessed May 14, 2015. https://doi.org/10.3168/jds.2011-4338
Sargolzaei M., Schenkel F.S., Jansen, G.B.& Schaeffer, L.R. 2008. Extent of linkage disequilibrium in Holstein cattle in North America, Journal of Dairy Science, 91, 2106–2117. https://doi.org/10.3168/jds.2007-0553
Sargolzaei, M. & Schenkel, F. S. 2009. QMSim: a large-scale genome simulator for livestock. Bioinformatics, 25, 680-681. https://doi.org/10.1093/bioinformatics/btp045
Sorensen, D. A. & Kennedy, B. W. 1984. Estimation of response to selection using least squares and mixed model methodology, Journal of Animal Science, 58, 1097–1103. https://doi.org/10.2527/jas1984.5851097x
Van der Werf, J. H. J., & de Boer, I. J. M. 1990. Estimation of additive genetic variance when base populations are selected, Journal of Animal Science, 68, 3124–3132. https://doi.org/10.2527/1990.68103124x.
Van Gestel, S., Houwing‐Duistermaat, J. J., Adolfsson, R., van Duijn, C. M., & van Broeckhoven, C. 2000. Power of selective genotyping in genetic association analyses of quantitative traits, Behavioral Genetics, 30, 141–146. https://doi.org/10.1023/a:1001907321955
Van Raden, P. M., Tooker, M. E., & Cole, J. B. 2009b. Can you believe those genomic evaluations for young bulls? Journal of Dairy Science, 92, 314 (Abstr.)
Van Raden, P. M., Van Tassell, C. P., Wiggans, G. R., Sonstegard, T. S., Schnabel, R. D., Taylor J. F., & Schenkel, F. S. 2009a. Invited review: Reliability of genomic predictions for North American Holstein bulls, Journal of Dairy Science, 92, 16–24. https://doi.org/10.3168/jds.2008-1514
Vitezica, Z. G., Aguilar, I., Misztal, I., &Legarra, A. 2011. Bias in genomic predictions for populations under selection, Genetics Research, 93, 357–366. https://doi.org/10.1017/S001667231100022X
Xu, S., &Vogl, C. 2000. Maximum likelihood analysis of quantitative trait loci under selective genotyping, Heredity, 84, 525–537. https://doi.org/10.1046/j.1365-2540.2000.00653.x
Yang, H., & G. Su. 2016. Impact of phenotypic information of previous generations and depth of pedigree on estimates of genetic parameters and breeding values, Livestock Science, 187, 61–67. doi:https://doi.org/10.1016/j.livsci.2016.03.001
Acknowledgements
Authors duly acknowledge the support provided by the Director ICAR-National Dairy Research Institute for providing the necessary facilities in the lab for carrying out the above work.
Author information
Authors and Affiliations
Contributions
GRG carried out the simulation and analysis and also prepared the manuscript draft. VV, AM, and RA helped in editing of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study did not require the ethics committee approval as no material derived from animals was used in this study.
Consent for publication
All authors gave their consent for the publication of the results obtained in this study.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Gowane, G.R., Alex, R., Mukherjee, A. et al. Impact and utility of shallow pedigree using single-step genomic BLUP for prediction of unbiased genomic breeding values. Trop Anim Health Prod 54, 339 (2022). https://doi.org/10.1007/s11250-022-03340-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11250-022-03340-2