
Abstract
Molecular scissors (MS), incl. Zinc Finger Nucleases (ZFN), Transcription-activator like endoncleases (TALENS) and meganucleases possess long recognition sites and are thus capable of cutting DNA in a very specific manner. These molecular scissors mediate targeted genetic alterations by enhancing the DNA mutation rate via induction of double-strand breaks at a predetermined genomic site. Compared to conventional homologous recombination based gene targeting, MS can increase the targeting rate 10,000-fold, and gene disruption via mutagenic DNA repair is stimulated at a similar frequency. The successful application of different MS has been shown in different organisms, including insects, amphibians, plants, nematodes, and mammals, including humans. Recently, another novel class of molecular scissors was described that uses RNAs to target a specific genomic site. The CRISPR/Cas9 system is capable of targeting even multiple genomic sites in one shot and thus could be superior to ZFNs or TALEN, especially by its easy design. MS can be successfully employed for improving the understanding of complex physiological systems, producing transgenic animals, incl. creating large animal models for human diseases, creating specific cell lines, and plants, and even for treating human genetic diseases. This review provides an update on molecular scissors, their underlying mechanism and focuses on new opportunities for generating genetically modified farm animals.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
With the exception of active transgenesis by vectors such as transposons and viral vectors, genetic modification starts with the creation of a double-strand break (DSB) in the DNA. The efficiency of a targeted genetic modification can be significantly enhanced by creation of a site-specific DSB (Rouet et al. 1994a). Molecular scissors normally consist of the cleavage domain and a DNA binding domain, which can be designed to bind to nearly any DNA sequence. By selecting for different outcomes of DNA repair, either gene knockout or targeted transgene insertion can be obtained. Here, we review the state of the art on different molecular scissors, highlight their underlying biological mechanism and provide milestone results that have been achieved by using the different MS categories. We start with the history of molecular scissors, then outline the strategies to modify a genome via MS, and finally survey the growing literature on the utility of MS for targeted genome modification.
Causing double-strand breaks stimulates targeted genetic modifications
Homologous recombination (HR) is a rare cellular event that has numerous applications, such as for studying basic mechanisms in mammalian development and physiology, and for producing transgenic farm animals that could play an important role in xenotransplantation, or will be used as human disease models, or for gene pharming or simply to increase breeding performance. In ES-cells, HR can be achieved using a positive–negative selection approach based on the presence of an antibiotic selection cassette within the homologous region which will confer resistance against an antibiotic drug. With a promoterless approach, the resistance cassette is driven by an endogenous active promoter, which significantly reduces the amount of false positive cell clones. This can be combined with a selection cassette localized outside of the homologous region. The combined approach reduces the amount of false positive selected cell clones. Using the yeast I-SceI meganuclease, it was shown that the DSB within the targeted region of the host genome is compatible with efficient HR by taking advantage of the cellular DNA repair mechanisms (Rouet et al. 1994b). Several studies have reported 1–18 % HR events per mammalian cell when the targeted DSB was introduced by natural or artificial endonucleases compared to 10−6 HR events without the use of endonucleases (Choulika et al. 1995; Donoho et al. 1998; Epinat et al. 2003; Vasquez et al. 2001; Szczepek et al. 2007; Urnov et al. 2005). Meganucleases were the first molecular scissors that were discovered and used to cut target DNA within the host genome. In the past few years, new molecular scissors were discovered that cut DNA in a very precise manner, with unprecedented efficiency and in a straightforward manner. These new molecular scissors include Zinc-Finger nucleases (ZFNs), Transcription-activator like endonucleases (TALEN) and the recently described RNA-programmable genome editors (CRISPR/CAS9). Synthetic endonuclease enzymes such as ZFNs, transcription activator-like effector nucleases (TALEN) and the emerged CRISPR/Cas9 are applicable to both direct (microinjection) and cell-mediated transgenesis and enable both gene inactivation by mutagenesis and precise sequence deletion, replacement or insertion by gene targeting and introduction of single nucleotide polymorphisms into the genome of species. A combination of several molecular scissors can also be used to delete large regions of genes and chromosomes. By using these precise and highly efficient technologies, researchers have now entered an era of “genome editing”.
Meganucleases
Structure of meganucleases
Meganucleases, also called homing endonucleases, can be divided into five families based on sequence and structure motifs: LAGLIDADG, GIY-YIG, HNH, His-Cys box and PD-(D/E)XK (Arnould et al. 2011; Silva et al. 2011; Orlowski et al. 2007). Meganucleases are generally encoded within introns or inteins although freestanding members also exist (Chevalier and Stoddard 2001). Meganucleases target long DNA sequences (14–40 bp) that are recognized and cleaved with high specificity in vitro and in vivo. By using such a large recognition sequence, meganucleases tolerate some target-site polymorphisms with little or no loss in binding and cleavage activity. Such long target sequences are rarely found in the genome, e.g. only one I-SceI target site can be found in the entire yeast genome. The best studied family of meganucleases are the LAGLIDADG proteins. The defining sequence motif, LAGLIDADG, represents the essential element for enzymatic activity. Some of the proteins contain only one of these motifs (I-CreI), others contain two motifs (PI-SceI). The defining sequence motif, LAGLIDADG, is essential for enzymatic activity. In single and double motif endonucleases, the domain adopted a similar αββαββα fold, with the LAGLIDADG motif comprising the terminal region of the first helix, not only contributing to a bipartite catalytic center but also forming the core subunit/subunit interaction. Two such α/β domains must assemble to form the functional protein, with the resulting β-strands creating a saddle-shaped DNA binding interface.
By mutating individual protein residues close to the DNA, the specificity of meganucleases can be altered without disturbing the catalytic efficiency (Rosen et al. 2006; Sussman et al. 2004; Seligman et al. 2002; Doyon et al. 2006). Different laboratories tested the concept of subunit inter-changeability by fusing both similar and disparate α/β domains, giving rise to hybrid proteins with specificity derived from each of the parental protein targets. These experiments revealed the feasibility of a “mix and match” approach, and confirmed the evolutionary concept that the double-motif LAGLIDADG proteins arose from a gene duplication event. Strategies for engineering meganucleases involve a semi-rational approach in which specific residues are mutated on the basis of prior structural or functional knowledge to create libraries with limited diversity. An automated high-throughput screening method was used for the I-CreI scaffold, resulting in the identification of hundreds of mutants with altered specificities (Arnould et al. 2006; Smith et al. 2006). Nevertheless, bona fide engineering of the specificity of meganucleases turned out to be more complex. Comprehensive computational studies gave rise not only to specificity re-engineering but also addressed dimerization via targeting protein–protein interactions of the subunits.
Gene targeting using meganucleases
DNA damage occurs naturally and frequently is associated with chromosomal aberrations. Normally, the cell activates its repair mechanisms to ensure genomic integrity. This repair mechanism can either be error-prone non-homologous end joining (NHEJ) or conservative (HR) depending on the mechanism involved.
I-SceI is the prototype of a meganuclease used for genome engineering. In nature, I-SceI stimulates HR by creating site-specific DSB in a process called homing. Discovery of this function in the yeast Saccharomyces cerevisiae paved the way into a new era in gene targeting (Jacquier and Dujon 1985). Pioneering experiments showing the usefulness of meganucleases for gene targeting were performed in the 1990s in which a chromosomal neomycin resistant gene interrupted by an I-SceI recognition site was used as a reporter to monitor gene correction events in mouse cell lines. Gene correction could be achieved at frequencies of 3 × 10−5 in ES cells and 4 × 10−4 in NIH3T3 cells, and up to 10 % recombination in HEK-293 cells using I-SceI (Smih et al. 1995; Rouet et al. 1994a); (Szczepek et al. 2007). Studies with the I-SceI system to decipher the cellular mechanisms of DSB repair have laid the foundation for the main strategies envisioned today for genome engineering. As cleavage sites for naturally occurring meganucleases do not exist in mammalian genomes, a cleavage site has to be introduced by transfection into the region of interest prior to use, which is an obvious drawback of natural meganucleases. Nevertheless, it has been successfully employed to induce various genetic modifications, incl. point mutations, recombination between repeats or gene targeting in bacteria (Horzempa et al. 2010; Yu et al. 2008; Flannagan et al. 2008; Posfai et al. 1999), mosquitos (Windbichler et al. 2007), flies (Maggert et al. 2008), and plants (Siebert and Puchta 2002; Puchta 2002, 1999). I-SceI was also used to improve efficiency of genetic modification in multiple organisms incl. frogs (Loeber et al. 2009), flies (Takeuchi et al. 2007), fish (Grabher and Wittbrodt 2008), and sea anemone (Renfer et al. 2010). The company Cellectis Bioresearch (Paris, France) distributes products and protocols for transgene integration into several types of cell lines, e.g. CHO-K1, CHO-S, NIH3T3 or HEK-293.
Re-engineered meganucleases have overcome the limitations of natural meganucleases and have been employed for targeting the RAG1 gene in 293 cells and resulted in a 6 % increase of targeted modifications (Grizot et al. 2009). Recently, an engineered I-CreI meganuclease was used for the first time to demonstrate the feasibility to generate knockout animals. Menoret et al. applied an engineered I-CreI meganuclease targeting the mouse and rat recombination activating gene 1 (RAG1) and injected the encoding plasmid into pronuclei of mouse and rat zygotes (Menoret et al. 2013). RAG1 is mainly expressed in T- and B-lymphocyte precursors and cleaves V(D)J T cell receptor and immunoglobulin DNA coding sequences to yield the large diversity of the immune cell repertoire (De and Rodgers 2004; de Villartay et al. 2003). Deficiency of the RAG1 gene results in severe combined immunodeficiency (SCID). Microinjection of the RAG1-meganuclease into mouse 1-cell embryos yielded high survival rates (67 %), similar to microinjection of conventional DNA. A mutation was detected in ~25 % of the fetuses (3.4 % of the microinjected embryos). In all five mouse fetuses with mutations, deletions ranged from 6 to 46 nt, three animals showed mutations in both alleles. Some pregnancies were allowed to go to term and one mutated founder animal was obtained among the newborn (4 % of newborn, 0.6 % of microinjected embryos). The mutated founder animal showed a 5-bp deletion due to the deletion of 8 bp of genomic DNA and insertion of 3 bp. The founder was bred to a wild-type animal and the mutation was transmitted to the offspring. Thereby, after the second round of breeding homozygous animals were obtained. These homozygous RAG1-KO rats showed reduced spleen, thymus and lymph node size compared to WT-controls. Additionally, the total number of cells from the primary lymphoid organs, the thymus and bone marrow were significantly reduced by 98.4 and 33.4 %. The number of cells in the secondary lymphoid organs, the spleen and lymph nodes were significantly reduced by 86 and 77 %, respectively. Overall, the animals showed a reduced number of TCR+ CD4+ and CD8+ cells in spleen and thymus. B-cell differentiation in the bone marrow revealed a significantly reduced percentage of pre-B and B-cells. Other immuno-active cells such as NK-cells were also reduced. Graft survival was significantly prolonged in RAG1-KO rats. These experiments showed for the first time the applicability of engineered meganucleases for genetic engineering of laboratory animals. This approach could be extended to farm animals in the future.
Zinc finger nucleases
Structure of zinc finger nucleases
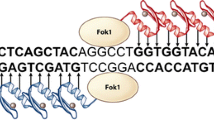
The first zinc-finger (ZF) protein has specific binding affinity to DNA and was discovered as part of the transcription factor IIIa in Xenopus oocytes (Miller et al. 1985). A typical zinc finger (Cys2His2) consists of 30 amino acids which form two anti-parallel β-sheets opposing an α-helix (Pabo et al. 2001). The domain is stabilized by two cysteine and two histidine residues binding a zinc ion, thus forming a compact globular domain. The zinc-finger motif uses residues in the alpha helix to bind to approximately 3 specific base pairs in the major groove of the DNA (Pavletich and Pabo 1991). ZFs can be designed for binding to almost any triplet (Pabo et al. 2001). Multiple ZFs can be combined to form a larger DNA-recognition domain which in turn increases specificity and efficiency of genetic modification. Specific binding of individual zinc fingers is largely independent, with some contacts between adjacent fingers altering base pair recognition. While the zinc-finger motif was discovered in the 1980s (Miller et al. 1985), ZFNs have a shorter history. The first specific ZFN was reported ~15 years after the discovery of the zinc-finger domain (Kim et al. 1996). A ZFN consists of a site-specific zinc-finger DNA binding domain fused to the nonspecific cleavage domain of the FokI endonuclease. At least, two ZFN-molecules are required for genetic modification, since the FokI nuclease must dimerize to cut the DNA, which doubles the number of specifically targeted base pairs (Smith et al. 2000). The two ZFN molecules bind to the targeted DNA in a tail-to-tail orientation separated by 5–7 bp, with double-stranded DNA cleavage occurring in the spacer region (Fig. 1).

Schematic drawing of a zinc-finger nuclease (ZFN) which binds as juxtaposed pairs. Zinc-finger (ZF) molecules bind to their specific DNA sequence. Each ZF molecule is specific to a base triplet. To enhance specificity 3–6 (or more) ZF molecules are linked to each other to recognize a DNA sequence of 9–18 bp. Dimerization of the FokI endonuclease causes a double strand break in the DNA
Modification of livestock genomes using zinc-finger nucleases
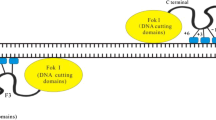
To employ a specific ZFN for genetic engineering, the plasmid DNA or mRNA encoding a specific ZFN is introduced into cells or embryos via microinjection or transfection [Fig. 4, see also (Hauschild-Quintern et al. 2013a)]. After translation, the ZFN pair binds to its specific target, the FokI nucleases dimerize and the DNA is cleaved. ZFN activity is enhanced by incubating transfected cells at 30 °C for a few days (Doyon et al. 2010). Cultivation at subphysiological temperatures slows down the cell cycle, giving the ZFNs more time to bind and cut at the targeted locus. A ZFN pair ideally induces a site-specific DSB only at the genomic site for which the molecule had been designed. Nevertheless, the possible threat of inducing off-target cleavage requires a comprehensive profiling of the putative off-target cleavage sites, which is necessary in every use of any molecular scissor.
After ZFN-mediated DNA cleavage in eukaryotic cells, DSB repair is initiated, either by NHEJ or HDR (Fig. 2). NHEJ is error-prone and often creates mutations of a few base pairs (average 10–20 bp) in the repaired chromosome (Bibikova et al. 2002). Such mutations can cause frame-shift or disruption of a gene, which in turn leads to the genetic knockout. Since the frequency of genetic modification is generally >1 %, isolation of knockout cells is readily achieved by interrogation of cell clones generated by limiting dilution. Fluorescence activated cell sorting (FACS) or magnetic bead selection have been successfully employed to enrich the targeted non-immortalized and other poorly clonable cells lines (Yu et al. 2011; Whyte et al. 2011; Hauschild et al. 2011).

Schematic drawing to demonstrate potential outcomes of a double-strand break in DNA generated by a pair of ZFNs, gene disruption or DNA insertion. If cells are treated with ZFNs alone, the repair process called non-homologous end-joining results in the rejoining of the broken ends of the DNA. As a consequence, insertions or deletions (indels) result in the generation of a shortened or non-functional protein (gene disruption). In contrast, if cells are treated with ZFNs in the presence of donor DNA that encodes for entire new gene or a small mutation of the endogenous gene, the cell can use the donor DNA as a template to repair the broken DNA. By using a coding sequence that is flanked by short arms homologous to the target sequence, a high frequency of targeted integration can be obtained. This process is called homology-directed repair (HDR)
Mitotic cells often repair a DSB using homology-based DNA repair. In such a case, the cell normally uses the sister chromosome as a template to repair the DSB. When a donor DNA molecule containing homologous arms to both sides of the DSB is co-transfected with the ZFNs, the molecule can be used as template. The exogenous DNA sequence placed between the two regions of homology will be copied into the chromosome during the DNA repair process (Moehle et al. 2007). In the absence of a site-specific break, the donor DNA must contain a large region (6–7 kb) homologous to the targeted region for capturing one of the rare spontaneous breaks (Deng and Capecchi 1992). In contrast, ZFN-based targeting strategy is compatible with a significantly shorter stretch of homologous DNA. Typically, 500–1,500 bp are used and even 50 base pairs on each side can be sufficient for site-specific integration (Orlando et al. 2010).
Only the sequence between the homologous regions is generally inserted into the genome, not the ZFN plasmid DNA. Integration of the ZFN DNA could result in constant transcription of the ZFN and would likely lead to non-specific DNA cleavage. Usually, ZFN plasmids are rapidly diluted and disappear from the treated cells when a transient transfection protocol is applied. This is a major advantage of ZFN mediated targeting, as the lack of integration prevents negative side effects such as insertional mutagenesis. Nevertheless, the transfection or microinjection of mRNA encoding for ZFN is a reasonable solution to further reduce the risk of integration and permanent expression of ZFNs (Watanabe et al. 2013). ZFNs have been broadly applied in basic research, biotechnology and medicine, but genome engineering with ZFNs is limited by the random generation of unwanted indels at homology sites (Liu et al. 2013). One additional potential strategy to overcome this limitation is the targeted introduction into DNA containing a single-strand break (SSB) or nick. A nick can be equivalent to a DSB and stimulate the HDR pathway (Meselson and Radding 1975; Radding 1982). In contrast to a DSB, a nick is not a bona fide substrate for repair by the NHEJ pathway. Thus, a targeted nick has the potential to restrict repair to the HDR pathway (Wang et al. 2012).
Pig
Transgenic farm animals, specifically the domestic pig, increasingly serve as model for human diseases. This is an important complementation to the laboratory mouse where it has been shown that the typical disease manifestation often does not fully mimic the human disease symptoms. Pigs share many genetic, anatomical and physiological features with humans, and have rapidly emerged as a suitable model for specific diseases, incl. cystic fibrosis, diabetes, cancer (Flisikowska et al. 2012; Luo et al. 2012) and several neurological disorders (Rogers et al. 2008; van den Heuvel et al. 2012; Mikkelsen et al. 1999). Pigs are also considered as suitable organ donors for xenotransplantation to reduce or even eliminate the shortage of suitable human organs (Cooper and Ayares 2011). This requires genetic modification of the donor pigs to overcome the severe immunological rejection responses after pig-to-primate xenotransplantation. Conventional HR targeting is extremely inefficient and normally does not lead to a bi allelic KO. Moreover, true pluripotent cells are not yet available from pigs and other domestic animals, preventing selection for rare HR events as in laboratory mice. The production of transgenic farm animals is significantly facilitated by effective somatic cell nuclear transfer (SCNT) protocols (Petersen et al. 2008). This cell-mediated transgenesis is compatible with screening for the genetic modification and analysis of the transgenic genotype in the laboratory rather than in animals ‘on the farm’. These cells are then used to produce the modified phenotype. While cell-mediated transgenesis is more labour intensive than direct transgenesis, in vitro genetic manipulation of cells followed by detailed genome analysis bears significant advantages. First, it reduces the total number of animals required to generate a useful transgenic offspring. Second, it increases dramatically the number of independent transgene integration events that can be screened and investigated. Third, it facilitates the engineering of precisely controlled genetic alterations (gene targeting) by allowing selection and isolation of rare integration events resulting from HR.
ZFN targeting of the transgenic eGFP (pCX-eGFP) locus in the domestic pig, with ~10 genomic integration sites, decreased fluorescence intensity due to mutation of some of the multiple ZFN targets. After targeting, the rate of non-fluorescent cells increased from 6 % (control) to 21 % (ZFN-targeted cells), showing that in ~15 % of the cells nearly all copies of the eGFP gene had been disrupted. Sequencing of several non-fluorescent cell clones revealed that wild-type DNA (non-mutated eGFP) variants remained, implying that at least one intact eGFP copy was silenced (Watanabe et al. 2010).
The first live ZFN mediated KO pig carried a hemizygous transgenic eGFP allele. Porcine fibroblasts were co-transfected with a pair of ZFN plasmids and a red-fluorescent CAG-tomato plasmid (transient selectable fluorophore). Two percent of the cells showed red fluorescence and could be sorted by FACS. A second round of selection for green cells by FACS led to 5 % eGFP negative cells. Selected cells used in SCNT led to the delivery of six out of seven piglets without the specific eGFP fluorescence. Sequencing revealed several deletions and insertions (Whyte et al. 2011). A third litter with six piglets was eGFP negative. One piglet had an unusual large deletion of 700 base pairs with nearly the entire eGFP coding sequence (Whyte and Prather 2011).
The first endogenous porcine gene successfully targeted by ZFN was the peroxisome proliferator-activated receptor-γ (Ppar-γ) locus. Ppar-γ −/− animals could be useful for studies on cardio-vascular diseases. Male fibroblasts were co-transfected with a Ppar-γ-specific ZFN pair and a neomycin-resistance gene. After selection with G418, 4 % of screened cell clones carried a mutated Ppar-γ gene and served as donors in SCNT. Two live piglets carried a mutation in one of the Ppar-γ alleles. Western blotting analysis confirmed the successful production of heterozygous Ppar-γ KO animals (Yang et al. 2011).
The first live pigs with bi-allelic KO via ZFN targeting of an endogenous gene were generated by our laboratory (Hauschild et al. 2011). Transfection of fetal fibroblasts with a pair of ZFN plasmids directed against exon 9 of the α1,3-galactosyltransferase (GGTA1, Gal) gene, induced —bi-allelic mutations 1 % of the cells. With the aid of magnetic beads, Gal-negative cells could be successfully enriched and >99 % of the cells were Gal-negative. After use of the selected cells in SCNT, Gal-negative fetuses were obtained 25 days after transfer of reconstructed embryos to recipients. In total, nine liveGGTA1-knockout piglets were produced from cloning with fetal fibroblasts. Sequencing revealed five different haplotypes with two homozygous and three heterozygous (individual mutations on each allele) mutations. The GGTA1 gene showed deletions from 1 to 7 base pairs in size and one large deletion of 96 bp. The GGTA1 KO fibroblasts derived from the ZFN approach were protected in against lysis in a complement in vitro assay to a similar extent as the existing HR-Gal-KO cells. Results demonstrate that homozygous GGTA1-KO pigs can be produced within 6 months which is significantly faster than with conventional HR. In a follow-up study, it was shown that the efficiency of the ZFNs is not influenced by the gender of the cells (Hauschild-Quintern et al. 2013b), thus allowing production of knockout pigs of both sexes with similar efficiency.
Cattle
In cattle, ZFN-mediated gene targeting was conducted to produce beta-lactoglobulin (BLG)-KO animals. BLG is the major whey protein in bovine milk and is the critical milk allergen. Bovine fetal fibroblasts were transfected with mRNA coding for ZFNs designed against the BLG gene. Sequencing revealed that ~15 % of the cells carried a mutated variant and 3 % of the single cell colonies showed a bi-allelic BLG-gene knockout. Homozygous KO-cells were used in SCNT and eight cloned animals were born; one survived the postnatal period. The mutated BLG gene was shorter (9 and 15 base pairs deletion, no frame shift) than the wild-type version. Off-target site mutations induced by the ZFN pair were also analyzed for BLG. While a one base pair mismatch with the targeting sequence led to 7 % gene targeting (single nucleotide polymorphism in cattle), three and seven base pair mismatches did not result in a mutated phenotype in sheep and pigs. Results suggest that ZFN-mediated targeting is promising for specific gene editing in large domestic animals with little risk of off-target site cleavage (Yu et al. 2011).
Transcription activator like effector endonucleases (TALEN)
Structur of TALENs
TALEs (transcription activator like effector) are naturally produced by plant pathogens such as Xanthomonas, a gram-negative bacteria, that can infect a wide variety of plant species including pepper, rice, citrus, cotton, tomato and soybeans (Boch et al. 2009; Boch and Bonas 2010). TALEs bind to their host DNA, act as transcription factors and and activate the expression of plant genes that aid bacterial infection. Plants have developed a defense mechanism against type III effectors that includes resistance-genes triggered by these effectors. Some of these genes appear to have evolved to contain TAL-effector binding sites similar to sites in the intended target genes. This competition between pathogenic bacteria and the host plant has been hypothesized to account for the malleability of the TAL effector DNA binding domain (Voytas and Joung 2009). TALEs consist of repeats, each consisting of 33–35 amino acids with two polymorphisms at positions 12 and 13 within the module, which are called the repeat variable di-residue (RVD). One RVD binds specifically to one nucleotide of genomic DNA (Moscou and Bogdanove 2009; Boch et al. 2009), hence establishing a 1:1 code for protein-DNA interaction (Fig. 3).

A schematic representation of a transcription activator-like effector nuclease (TALEN) pair is shown. Each TALEN is composed of transcription activator-like effectors (TALEs) at the amino terminus and the FokI nuclease domain at the carboxyl terminus. Each TALE repeat consists of 33–35 amino acids and recognizes a single base pair through the amino acids at positions 12 and 13, which is called the repeat variable residue (RVD). Target sequences are typically 30–40 bp in length
Individual TALE repeats can be used to engineer DNA binding domains capable of recognizing endogenous sequences in mammalian cells. By linking the binding domain with the non-specific cleavage domain from the type IIs restriction endonuclease FokI, TALENs can be used as tool for stimulating NHEJ and HR (Cermak et al. 2011; Christian et al. 2010; Li et al. 2011a, b; Mahfouz et al. 2011; Miller et al. 2011). Given the modular nature of this DNA binding domain, RVDs with different specificities can be assembled into arrays in order to target user-defined DNA sequences.
TALENs can be successfully used to target endogenous genes and efficiently cleave DNA leading to NHEJ (Hockemeyer et al. 2011). A comparative study with human ES-cells and induced pluripotent stem cells and three different target genes AAVS1, OCT4 and PITX3 revealed that TALENs and ZFNs had a similar targeting efficiency (Hockemeyer et al. 2011). TALENs have been used to knockout genes in rats and zebrafish (Tesson et al. 2011; Sander et al. 2011; Huang et al. 2011) and in cattle, sheep and pigs, thus demonstrating that TALENs are effective in inducing genetic modifications in a broad range of different species (Carlson et al. 2012; Proudfoot et al. 2015).
ZFNs and TALENs differ in three main aspects: (1) TALE repeats are 3–4 times larger than ZFNs, when recognized per base pair of the targeted DNA. This may interfere with viral delivery methods, particularly adeno-associated virus; (2) the spacer length (the gap between two binding sites) is variable and not restricted to a specific length, which complicates TALEN design and could lead to greater off-target activity relative to an identical nuclease with a fixed spacer length, (3) ZFNs assembly requires an archive of high-quality, well characterized modules to achieve specific gene targeting because cross-talk between the individual fingers can lead to imperfect DNA recognition (Defrancesco 2011). Context-dependent effects between the repeat units, as reported for ZFNs (Cathomen and Joung 2008), have not been reported so far for TALENs.
Various assembly methods have indicated that TALE repeats can be combined to recognize potentially any target sequence, the only limitation is that TALE binding sites must start with thymidine (Boch and Bonas 2010). This needs to be considered when screening a locus for potential target sites. TALENs appear to be superior to ZFNs in terms of simplicity and straightforwardness in design and assembly strategies. Manufacture of effective TALENs is cheaper and faster compared to ZFNs. The relative simple TALE assembly is displayed in a recent study reporting the construction of a library of TALENs targeting 18,740 different human protein-coding genes (Kim et al. 2013). Active, custom-designed TALENs have been reported to induce indel frequencies between 2 and 55 % of targeted chromosomes (Carlson et al. 2012). TALENs can be easily designed and assembled using molecular biology techniques available in most laboratories around the world.
Modification of livestock genomes using TALENs
Cattle
Carlson et al. (2012) designed TALENs to target the bovine ACAN and GDF8 genes in fibroblasts. ACAN, also known as Aggrecan is thought to play an important role in the formation of congenital achondroplasia, while GDF8 (growth differentiation factor 8, myostatin) is a regulator of muscle growth. A non-functional myostatin gene is known to cause muscular hypertrophy in Belgian Blue and Piedmontese cattle. GDF8-targeted bovine fibroblasts showed a modification of the gene in 7 out of 24 cell clones (29 %). None of the cell clones carried a bi-allelic modification. The ACAN gene was targeted in 27 out of 35 cell clones (77 %). Two cell clones showed a bi-allelic modification. Modified cells could be used as donor cells for somatic cell nuclear transfer to produce live offspring carrying the desired genetic modification (Carlson et al. 2012).
Pig
The LDL receptor gene was targeted with the aid of TALEN in pigs to create a model for familial hypercholesterolemia (Carlson et al. 2012) and the porcine DMD gene to create a model for Duchenne Muscular Dystrophy. The most active TALEN pair targeting the DMD gene had a cleavage efficiency of 38 %. The DMD gene was successfully targeted in 41 % of analysed cell clones with ~30 % of these carrying a bi-allelic mutation. The combined transfection of TALEN pairs targeting exons six and seven of the DMD locus resulted in deletion of 6.5 kb DNA in 10.3 % of selected colonies. Mono- and bi-allelic LDLR gene modified cell clones were pooled and used as donors for somatic cell nuclear transfer. Pregnancies were established after seven of nine transfers. Six pregnancies were maintained to term, and yielded 18 live-born piglets of which eight contained mono-allelic mutations and ten contained bi-allelic mutations of the LDLR gene. To enhance disease resistance in pigs, 20 ng/μL specific TALEN mRNA were microinjected into porcine zygotes to target the porcine RELA gene (p65) which is critically involved in tolerance against African Swine Fever virus infection (Palgrave et al. 2011). Sixteen out of 56 successfully injected embryos revealed indels detected by Surveyor assay and/or sequence analysis. One-third of the mutants were either homozygous or heterozygous mutants. RELA-mutated porcine embryos were not transferred to assess in vivo development.
These results demonstrate the robustness and reproducibility of TALEN to nearly any genomic locus for which the genomic sequence is available. These results clearly show that TALENs are compatible not only with the deletion at a defined genomic locus, but also allow precise allelic introgression and large chromosomal deletions/inversions, rendering TALEN a valuable tool for genetic modification of farm animals.
Sheep
Proudfoot et al. (2015) recently described the generation of gene edited sheep. As the bovine and ovine genomic sequence of the MSTN locus showed high similarity between both species, they used the same TALENs that successfully targeted the bovine MSTN locus. Transient transfection of TALEN mRNA into ovine cells and subsequent analysis by the Surveyor nuclease assay showed similar levels of activity in both species. To generate living offspring, they microinjected TALEN mRNA in in vitro produced ovine zygotes and transferred blastocysts to synchronized recipient ewes. In total, 26 blastocysts were transferred to 9 recipients resulting in 8 pregnancies and 12 live births. One of the offspring carried a heterozygous gene editing in the targeted locus. This study further exemplifies the utility and ease with which TALENs can be used to engineer the genome of livestock (Proudfoot et al. 2015).
RNA guided genomic engineering (CRISPR/Cas9)
Structure of CRISPR/CAS9
The CRISPR/Cas9 system has recently emerged as potentially facile and efficient alternative to ZFNs, TALENs and other meganucleases for inducing targeted genetic alterations, and has revolutionized the field for targeted genomic engineering in the short time since its appearance. In bacteria and archaea, CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)/Cas (CRISPR-associated) loci encode RNA-guided adaptive immune systems that can destroy foreign DNA (Bhaya et al. 2011; Terns and Terns 2011; Wiedenheft et al. 2012). The Streptococcus pyogenes SF370 type II CRISPR locus consists of four genes, including the Cas9 nuclease and two non-coding RNAs. TracrRNA and a pre-crRNA array containing nuclease guided sequences interspaced by identical direct repeats (Cong et al. 2013). In vitro reconstitution of the S. pyogenes CRISPR system demonstrated that crRNA fused to a normally trans-coded tracrRNA is sufficient to direct Cas9 protein to highly specific cleavage of target DNA sequences matching the crRNA (Mali et al. 2013). This redesign as a single transcript [single-guide RNA or guide RNA (gRNA)] encompasses the features required for both Cas9 binding and DNA target site recognition. Using sgRNA, Cas 9 can be programmed to cleave double-stranded DNA at any genomic site defined by the guide RNA sequence and a protospacer adjacent motif (PAM). The PAM is an essential targeting component that also serves as a self versus non-self recognition system to prevent the CRISPR locus itself from being targeted. Many type II systems have different PAM requirements, which may affect their usefulness and targeting efficiency. The most commonly engineered system, from Streptococcus pyogenes, requires a NGG protospacer adjacent motif (PAM), where N can be any nucleotide. In bacterial systems CRISPR/Cas can be used as it is, while in humans it involves expression of a human-codon-optimized Cas9 protein with an appropriate nuclear localization signal. Moreover, the crRNA and tracrRNA must be expressed either individually or as a single chimera via a RNA polymerase III promoter (Cong et al. 2013; Mali et al. 2013; Jinek et al. 2013). The typical features of CRISPR/Cas9 suggest that is a simple and versatile system for generating double-stranded breaks that facilitate site-specific genome editing. Moreover, CRISPR/Cas can target multiple loci by the sgRNA, potentially allowing simultaneous targeting of multiple genomic loci. CRISPR/Cas9 vectors are commercially available and can be used after introducing the specific gRNA sequence, which is a nucleotide of 20–30 bp (Fig. 4). The number of reports since the first description of the successful use of CRISPR/Cas for targeting a specific genomic locus has dramatically increased. Current data suggest that CRISPRs have similar specificity and efficiency as ZFNs and TALENs. In addition, CRISPRs have the advantage of being simply to generate, easy to handle, efficient and cost-effective. Open questions regarding their specificity have further to be addressed in future experiments. CRISPR-vectors with Nickase activity, to avoid off-target events (Shen et al. 2014), or vectors that have an inactivated version of the Cas-motif connected to the FokI endonuclease, which has to dimerize before cutting and thereby increases the specificity (Tsai et al. 2014; Guilinger et al. 2014), are already available. The specificity can be further enhanced by the use of a truncated gRNA (Fu et al. 2014).

Schematic representation of RNA-guided engineered nucleases are shown. In this example the molecule consists of the CRISPR (clustered regularly interspaced short palindromic repeat)—associated protein 9 (Cas9) and a single-chain guide RNA (sgRNA). The guide sequence in the sgRNA (or short gRNA) is complementary to a 20-bp target DNA known as protospacer, which is next to a protospacer adjacent motif (PAM). The PAM usually is a NGG sequence, where N represents any nucleotide
Application of CRISPR/Cas in domestic animals
Since the first description of the successful use of CRISPR/Cas for targeting a specific genomic locus, the number of reports has dramatically increased. Current, still preliminary data suggest that CRISPRs have similar specificity and efficiency as ZFNs and TALENs. In addition, CRISPRs have the advantage of being relatively simple to generate, easy to handle, and efficient and cost-effective. Open questions regarding their specificity have to be addressed in future experiments. CRISPR-vectors with Nickase activity to avoid off-target events are already available. First reports to the use of CRISPRs to modify the genomes of farm animals have been published (Hai et al. 2014; Li et al. 2014; Whitworth et al. 2014b; Tan et al. 2013).
Hai et al. (2014) targeted Exon 5 of the vWF (von Willebrand Factor) gene by injecting Cas 9 mRNA and sgRNA into porcine zygotes (Hai et al. 2014). In vitro development of the injected embryos did not differ from that of the control embryos, indicating that the Cas9 mRNA/sgRNA injection did not interfere with early embryonic development. A total of 76 injected embryos were transferred into 5 surrogate sows; 3 pregnancies were established, 16 piglets were delivered, 10 of these contained indels in the targeted site. Six piglets carried a bi-allelic knockout of the vWF gene. The survival rate of the piglets was 88 %, indicating that the treatment did not interfere with pig embryonic and fetal development in vivo. Expression of vWF at the protein level was not detectable in bi-allelic vWF-knockout pigs and the vWF KO pigs displayed a phenotype comparable to von Willebrand disease. Off-target cleavage events were not found.
CRISPR/Cas9 was also successfully employed to target the porcine p65-locus in fetal fibroblasts (Tan et al. 2013). Despite efficient production of DSBs in the target site, the frequency of CRISPR/Cas9 mediated HR was lower than with TALENs. Targeting the porcine APC-gene, CRISPR/Cas9 worked more efficiently, but still did not reach the level of HR induced by TALENs at the same site (30 vs. 60 %). These first reports on CRISPR/Cas suggest locus specific differences with regard to HR efficiency which may be adjusted by modifying the conditions under which CRISPR/Cas works best. There is still a controversial debate within the scientific community with regard to efficiency and precision of cleavage of ZFNs, TALENs and CRISPR/Cas. This may mainly be grounded on the different expertise the labs have with one or the other programmable endonuclease. The next generation of CRISPR/Cas systems is currently underway and will likely include, CRISPR/Cas with Nickase activity to further reduce the likelihood of off-target cleavage events and to increase the frequency of HDR. More studies are required in order to fully exploit the potential of CRISPR/Cas9.
Concluding remarks and future directions
Molecular scissors such as ZFNs, TALENs, and RNA-guided DNA endonucleases have emerged as valuable molecular tools that have the potential to revolutionize biological research with great benefits for personalized medicine. These emerging technologies significantly expand the ability to study model organisms, including large animals, and will be instrumental for correcting many genetic diseases. With the aid of these tools, researchers are able to develop biomedical models in species that are physiologically closer related to humans than mice. The domestic pig is particularly promising in this regard. The growing number of human disease models in pigs, supports this assumption (Flisikowska et al. 2014).
Due to the high degree of physiological similarity with humans, porcine organs are considered to be promising to combat the growing demand of human organs for allotransplantation. To achieve this goal and to avoid immune rejection responses, the pig genome has to be modified to ensure long term survival of porcine organs in patients after xenografting. ZFNs, TALENs and CRISPR/Cas can now be used to elegantly knockout porcine genes or to precisely knock-in transgenes at specific genomic sites in the porcine genome to produce pigs specifically tailored as organ donors.
However, to exploit the full potential of these new technologies, important questions and challenges must be addressed. A high degree of specificity is a main challenge and would be a critical prerequisite for employing these technologies in human patients. Comprehensive profiling of off-target cleavage sites will provide insight into the stringency of target recognition in each system, which in turn will help to increasing the specificity of the systems and to develop algorithms that calculate the most promising sequences to be targeted within a specific locus. Although CRISPR/Cas seems to show the greatest promise and flexibility for genetic engineering, sequence requirements within the PAM sequence may constrain some applications. Therefore, evolution of the Cas9 protein should pave the way towards PAM independence, and may also provide means to generate an even more efficient Cas9 endonuclease. Additional studies will also be required to evaluate the specificity and toxicity of RNA-guided DNA endonucleases in vitro and in vivo. Recent developments, in which an inactivated Cas element was conjugated to the FokI endonuclease, that requires dimer formation is promising as thereby a higher specificity can be achieved (Tsai et al. 2014; Guilinger et al. 2014). Biophysical and biochemical studies on CRISPRs could help to improve the design of next-generation genome editing tools.
The different nuclease systems have all their individual advantages and disadvantages (Table 1) and the selection of a specific system seems more to depend on the expertise of the individual researcher rather than on the weaknesses of one of these technologies . Until recently, somatic cell nuclear transfer (SCNT) was the only method of achieving gene targeting in large mammals. Nuclear transfer requires relatively few experimental animals as oocytes are obtained from animals slaughtered for other purposes. This is mainly true for species in which embryos at the zygote stage can not easily be obtained by flushing due to anatomical characteristics (pig). Still, SCNT remains the method of choice for many applications (Kurome et al. 2013), however, application of SCNT requires a high-level of technical expertise, reliable supply of oocytes and a large recipient herd, features not available in many areas where gene-editing might have the greatest impact (Proudfoot et al. 2015). With the emerge of the new highly efficient methods for transgenesis and genome editing, which makes it now feasible to generate genetically modified livestock by microinjection even in the absence of embryonic stem cells or true induced pluripotent stem cells and without the need of SCNT, we have now entered a new era, in which researchers can choose which method is the best for their purpose and to achieve their goals.
Gene knockout by the introduction of genome editing tool mRNA directly into early embryos offers a one-step method of gene inactivation without any cell intermediate, as shown for zebrafish (Doyon et al. 2008; Meng et al. 2008), rats (Geurts et al. 2009; Cui et al. 2011); mice (Carbery et al. 2010; Meyer et al. 2010; Wang et al. 2013; Yang et al. 2013), rabbits (Flisikowska et al. 2011), pigs (Hai et al. 2014; Whitworth et al. 2014a; Lillico et al. 2013), sheep and cattle (Tan et al. 2013; Proudfoot et al. 2015).
The advent of highly efficient genome editing tools has led to a renaissance in livestock genetic modification by embryo microinjection (Tan et al. 2013; Lillico et al. 2013). In comparison to SCNT, cytoplasmic injection of zygotes with mRNA of molecular scissors is both technically simple and efficient (Geurts et al. 2009; Carlson et al. 2012). Genome edited livestock differ from traditional genetically modified (GM) animals in that no recombinant DNA is integrated into the animal genome. Combined with the ability to mimic desirable or preexisting mutations by integration of a single nucleotide polymorphism (SNP), genome editing overcomes many of the issues associated with GM animals and can be seen as an adjunct technology to current breeding strategies to transfer desired traits into livestock. The ease and efficiency of genome editing tools lead to the fact that many laboratories worldwide can now contribute to the generation of genetically modified livestock, increasing the number of available animal models at a dramatic speed.
In summary, meganucleases, ZFNs, TALENs and CRISPRs are valuable tools, scientists 10 years ago could only dream of. The new tools significantly expand and revolutionize our ability to explore and alter any genome and constitute a new and promising paradigm to understand and treat genetic diseases and develop agricultural applications.
References
Arnould S, Chames P, Perez C, Lacroix E, Duclert A, Epinat JC, Stricher F, Petit AS, Patin A, Guillier S, Rolland S, Prieto J, Blanco FJ, Bravo J, Montoya G, Serrano L, Duchateau P, Paques F (2006) Engineering of large numbers of highly specific homing endonucleases that induce recombination on novel DNA targets. J Mol Biol 355(3):443–458. doi:10.1016/j.jmb.2005.10.065
Arnould S, Delenda C, Grizot S, Desseaux C, Paques F, Silva GH, Smith J (2011) The I-CreI meganuclease and its engineered derivatives: applications from cell modification to gene therapy. Protein Eng Des Sel PEDS 24(1–2):27–31. doi:10.1093/protein/gzq083
Bhaya D, Davison M, Barrangou R (2011) CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu Rev Genet 45:273–297. doi:10.1146/annurev-genet-110410-132430
Bibikova M, Golic M, Golic KG, Carroll D (2002) Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics 161(3):1169–1175
Boch J, Bonas U (2010) Xanthomonas AvrBs3 family-type III effectors: discovery and function. Annu Rev Phytopathol 48:419–436. doi:10.1146/annurev-phyto-080508-081936
Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, Lahaye T, Nickstadt A, Bonas U (2009) Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326(5959):1509–1512. doi:10.1126/science.1178811
Carbery ID, Ji D, Harrington A, Brown V, Weinstein EJ, Liaw L, Cui X (2010) Targeted genome modification in mice using zinc-finger nucleases. Genetics 186(2):451–459. doi:10.1534/genetics.110.117002
Carlson DF, Tan W, Lillico SG, Stverakova D, Proudfoot C, Christian M, Voytas DF, Long CR, Whitelaw CB, Fahrenkrug SC (2012) Efficient TALEN-mediated gene knockout in livestock. Proc Natl Acad Sci USA 109(43):17382–17387. doi:10.1073/pnas.1211446109
Cathomen T, Joung JK (2008) Zinc-finger nucleases: the next generation emerges. Mol Ther 16(7):1200–1207. doi:10.1038/mt.2008.114
Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, Baller JA, Somia NV, Bogdanove AJ, Voytas DF (2011) Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res 39(12):e82. doi:10.1093/nar/gkr218
Chevalier BS, Stoddard BL (2001) Homing endonucleases: structural and functional insight into the catalysts of intron/intein mobility. Nucleic Acids Res 29(18):3757–3774
Choulika A, Perrin A, Dujon B, Nicolas JF (1995) Induction of homologous recombination in mammalian chromosomes by using the I-SceI system of Saccharomyces cerevisiae. Mol Cell Biol 15(4):1968–1973
Christian M, Cermak T, Doyle E, Schmidt C, Zhang F, Hummel A, Bogdanove A, Voytas D (2010) Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 186(2):757–761
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339(6121):819–823. doi:10.1126/science.1231143
Cooper DK, Ayares D (2011) The immense potential of xenotransplantation in surgery. Int J Surg 9(2):122–129. doi:10.1016/j.ijsu.2010.11.002
Cui X, Ji D, Fisher DA, Wu Y, Briner DM, Weinstein EJ (2011) Targeted integration in rat and mouse embryos with zinc-finger nucleases. Nat Biotechnol 29(1):64–67. doi:10.1038/nbt.1731
De P, Rodgers KK (2004) Putting the pieces together: identification and characterization of structural domains in the V(D)J recombination protein RAG1. Immunol Rev 200:70–82. doi:10.1111/j.0105-2896.2004.00154.x
de Villartay JP, Fischer A, Durandy A (2003) The mechanisms of immune diversification and their disorders. Nat Rev Immunol 3(12):962–972. doi:10.1038/nri1247
Defrancesco L (2011) Move over ZFNs. Nat Biotechnol 29(8):681–684
Deng C, Capecchi MR (1992) Reexamination of gene targeting frequency as a function of the extent of homology between the targeting vector and the target locus. Mol Cell Biol 12(8):3365–3371
Donoho G, Jasin M, Berg P (1998) Analysis of gene targeting and intrachromosomal homologous recombination stimulated by genomic double-strand breaks in mouse embryonic stem cells. Mol Cell Biol 18(7):4070–4078
Doyon JB, Pattanayak V, Meyer CB, Liu DR (2006) Directed evolution and substrate specificity profile of homing endonuclease I-SceI. J Am Chem Soc 128(7):2477–2484. doi:10.1021/ja057519l
Doyon Y, McCammon JM, Miller JC, Faraji F, Ngo C, Katibah GE, Amora R, Hocking TD, Zhang L, Rebar EJ, Gregory PD, Urnov FD, Amacher SL (2008) Heritable targeted gene disruption in zebrafish using designed zinc-finger nucleases. Nat Biotechnol 26(6):702–708. doi:10.1038/nbt1409
Doyon Y, Choi VM, Xia DF, Vo TD, Gregory PD, Holmes MC (2010) Transient cold shock enhances zinc-finger nuclease-mediated gene disruption. Nat Methods 7(6):459–460
Epinat JC, Arnould S, Chames P, Rochaix P, Desfontaines D, Puzin C, Patin A, Zanghellini A, Paques F, Lacroix E (2003) A novel engineered meganuclease induces homologous recombination in yeast and mammalian cells. Nucleic Acids Res 31(11):2952–2962
Flannagan RS, Linn T, Valvano MA (2008) A system for the construction of targeted unmarked gene deletions in the genus Burkholderia. Environ Microbiol 10(6):1652–1660. doi:10.1111/j.1462-2920.2008.01576.x
Flisikowska T, Thorey IS, Offner S, Ros F, Lifke V, Zeitler B, Rottmann O, Vincent A, Zhang L, Jenkins S, Niersbach H, Kind AJ, Gregory PD, Schnieke AE, Platzer J (2011) Efficient immunoglobulin gene disruption and targeted replacement in rabbit using zinc finger nucleases. PLoS ONE 6(6):e21045. doi:10.1371/journal.pone.0021045
Flisikowska T, Merkl C, Landmann M, Eser S, Rezaei N, Cui X, Kurome M, Zakhartchenko V, Kessler B, Wieland H, Rottmann O, Schmid RM, Schneider G, Kind A, Wolf E, Saur D, Schnieke A (2012) A porcine model of familial adenomatous polyposis. Gastroenterology 143(5):1173–1175. doi:10.1053/j.gastro.2012.07.110
Flisikowska T, Kind A, Schnieke A (2014) Genetically modified pigs to model human diseases. J Appl Genet 55(1):53–64. doi:10.1007/s13353-013-0182-9
Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK (2014) Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol 32(3):279–284. doi:10.1038/nbt.2808
Geurts AM, Cost GJ, Freyvert Y, Zeitler B, Miller JC, Choi VM, Jenkins SS, Wood A, Cui X, Meng X, Vincent A, Lam S, Michalkiewicz M, Schilling R, Foeckler J, Kalloway S, Weiler H, Menoret S, Anegon I, Davis GD, Zhang L, Rebar EJ, Gregory PD, Urnov FD, Jacob HJ, Buelow R (2009) Knockout rats via embryo microinjection of zinc-finger nucleases. Science 325(5939):433. doi:10.1126/science.1172447
Grabher C, Wittbrodt J (2008) Recent advances in meganuclease-and transposon-mediated transgenesis of medaka and zebrafish. Methods Mol Biol (Clifton, NJ) 461:521–539. doi:10.1007/978-1-60327-483-8_36
Grizot S, Smith J, Daboussi F, Prieto J, Redondo P, Merino N, Villate M, Thomas S, Lemaire L, Montoya G, Blanco FJ, Paques F, Duchateau P (2009) Efficient targeting of a SCID gene by an engineered single-chain homing endonuclease. Nucleic Acids Res 37(16):5405–5419. doi:10.1093/nar/gkp548
Guilinger JP, Thompson DB, Liu DR (2014) Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat Biotechnol. doi:10.1038/nbt.2909
Hai T, Teng F, Guo R, Li W, Zhou Q (2014) One-step generation of knockout pigs by zygote injection of CRISPR/Cas system. Cell Res 24(3):372–375. doi:10.1038/cr.2014.11
Hauschild J, Petersen B, Santiago Y, Queisser AL, Carnwath JW, Lucas-Hahn A, Zhang L, Meng X, Gregory PD, Schwinzer R, Cost GJ, Niemann H (2011) Efficient generation of a biallelic knockout in pigs using zinc-finger nucleases. Proc Natl Acad Sci USA 108(29):12013–12017. doi:10.1073/pnas.1106422108
Hauschild-Quintern J, Petersen B, Cost GJ, Niemann H (2013a) Gene knockout and knockin by zinc-finger nucleases: current status and perspectives. Cell Mol Life Sci 70(16):2969–2983. doi:10.1007/s00018-012-1204-1
Hauschild-Quintern J, Petersen B, Queisser AL, Lucas-Hahn A, Schwinzer R, Niemann H (2013b) Gender non-specific efficacy of ZFN mediated gene targeting in pigs. Transgenic Res 22(1):1–3. doi:10.1007/s11248-012-9647-6
Hockemeyer D, Wang H, Kiani S, Lai CS, Gao Q, Cassady JP, Cost GJ, Zhang L, Santiago Y, Miller JC, Zeitler B, Cherone JM, Meng X, Hinkley SJ, Rebar EJ, Gregory PD, Urnov FD, Jaenisch R (2011) Genetic engineering of human pluripotent cells using TALE nucleases. Nat Biotechnol 29(8):731–734. doi:10.1038/nbt.1927
Horzempa J, Shanks RM, Brown MJ, Russo BC, O’Dee DM, Nau GJ (2010) Utilization of an unstable plasmid and the I-SceI endonuclease to generate routine markerless deletion mutants in Francisella tularensis. J Microbiol Methods 80(1):106–108. doi:10.1016/j.mimet.2009.10.013
Huang P, Xiao A, Zhou M, Zhu Z, Lin S, Zhang B (2011) Heritable gene targeting in zebrafish using customized TALENs. Nat Biotechnol 29(8):699–700. doi:10.1038/nbt.1939
Jacquier A, Dujon B (1985) An intron-encoded protein is active in a gene conversion process that spreads an intron into a mitochondrial gene. Cell 41(2):383–394
Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J (2013) RNA-programmed genome editing in human cells. Elife 2:e00471. doi:10.7554/eLife.00471
Kim YG, Cha J, Chandrasegaran S (1996) Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci USA 93(3):1156–1160
Kim Y, Kweon J, Kim A, Chon JK, Yoo JY, Kim HJ, Kim S, Lee C, Jeong E, Chung E, Kim D, Lee MS, Go EM, Song HJ, Kim H, Cho N, Bang D, Kim S, Kim JS (2013) A library of TAL effector nucleases spanning the human genome. Nat Biotechnol 31(3):251–258. doi:10.1038/nbt.2517
Kurome M, Geistlinger L, Kessler B, Zakhartchenko V, Klymiuk N, Wuensch A, Richter A, Baehr A, Kraehe K, Burkhardt K, Flisikowski K, Flisikowska T, Merkl C, Landmann M, Durkovic M, Tschukes A, Kraner S, Schindelhauer D, Petri T, Kind A, Nagashima H, Schnieke A, Zimmer R, Wolf E (2013) Factors influencing the efficiency of generating genetically engineered pigs by nuclear transfer: multi-factorial analysis of a large data set. BMC Biotechnol 13:43. doi:10.1186/1472-6750-13-43
Li T, Huang S, Jiang WZ, Wright D, Spalding MH, Weeks DP, Yang B (2011a) TAL nucleases (TALNs): hybrid proteins composed of TAL effectors and FokI DNA-cleavage domain. Nucleic Acids Res 39(1):359–372. doi:10.1093/nar/gkq704
Li T, Huang S, Zhao X, Wright DA, Carpenter S, Spalding MH, Weeks DP, Yang B (2011b) Modularly assembled designer TAL effector nucleases for targeted gene knockout and gene replacement in eukaryotes. Nucleic Acids Res 39(14):6315–6325. doi:10.1093/nar/gkr188
Li P, Estrada JL, Burlak C, Montgomery J, Butler JR, Santos RM, Wang ZY, Paris LL, Blankenship RL, Downey SM, Tector M, Tector AJ (2014) Efficient generation of genetically distinct pigs in a single pregnancy using multiplexed single-guide RNA and carbohydrate selection. Xenotransplantation. doi:10.1111/xen.12131
Lillico SG, Proudfoot C, Carlson DF, Stverakova D, Neil C, Blain C, King TJ, Ritchie WA, Tan W, Mileham AJ, McLaren DG, Fahrenkrug SC, Whitelaw CB (2013) Live pigs produced from genome edited zygotes. Sci Rep 3:2847. doi:10.1038/srep02847
Liu X, Wang Y, Guo W, Chang B, Liu J, Guo Z, Quan F, Zhang Y (2013) Zinc-finger nickase-mediated insertion of the lysostaphin gene into the beta-casein locus in cloned cows. Nat Commun 4:2565. doi:10.1038/ncomms3565
Loeber J, Pan FC, Pieler T (2009) Generation of transgenic frogs. Methods Mol Biol (Clifton NJ 561:65–72. doi:10.1007/978-1-60327-019-9_4
Luo Y, Bolund L, Sorensen CB (2012) Pig gene knockout by rAAV-mediated homologous recombination: comparison of BRCA1 gene knockout efficiency in Yucatan and Gottingen fibroblasts with slightly different target sequences. Transgenic Res 21(3):671–676. doi:10.1007/s11248-011-9563-1
Maggert KA, Gong WJ, Golic KG (2008) Methods for homologous recombination in Drosophila. Methods Mol Biol (Clifton NJ) 420:155–174. doi:10.1007/978-1-59745-583-1_9
Mahfouz MM, Li L, Shamimuzzaman M, Wibowo A, Fang X, Zhu JK (2011) De novo-engineered transcription activator-like effector (TALE) hybrid nuclease with novel DNA binding specificity creates double-strand breaks. Proc Natl Acad Sci USA 108(6):2623–2628. doi:10.1073/pnas.1019533108
Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM (2013) RNA-guided human genome engineering via Cas9. Science 339(6121):823–826. doi:10.1126/science.1232033
Meng X, Noyes MB, Zhu LJ, Lawson ND, Wolfe SA (2008) Targeted gene inactivation in zebrafish using engineered zinc-finger nucleases. Nat Biotechnol 26(6):695–701. doi:10.1038/nbt1398
Menoret S, Fontaniere S, Jantz D, Tesson L, Thinard R, Remy S, Usal C, Ouisse LH, Fraichard A, Anegon I (2013) Generation of Rag1-knockout immunodeficient rats and mice using engineered meganucleases. FASEB J 27(2):703–711. doi:10.1096/fj.12-219907
Meselson MS, Radding CM (1975) A general model for genetic recombination. Proc Natl Acad Sci USA 72(1):358–361
Meyer M, de Angelis MH, Wurst W, Kuhn R (2010) Gene targeting by homologous recombination in mouse zygotes mediated by zinc-finger nucleases. Proc Natl Acad Sci USA 107(34):15022–15026. doi:10.1073/pnas.1009424107
Mikkelsen M, Moller A, Jensen LH, Pedersen A, Harajehi JB, Pakkenberg H (1999) MPTP-induced Parkinsonism in minipigs: a behavioral, biochemical, and histological study. Neurotoxicol Teratol 21(2):169–175
Miller J, McLachlan AD, Klug A (1985) Repetitive zinc-binding domains in the protein transcription factor IIIA from Xenopus oocytes. EMBO J 4(6):1609–1614
Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, Meng X, Paschon DE, Leung E, Hinkley SJ, Dulay GP, Hua KL, Ankoudinova I, Cost GJ, Urnov FD, Zhang HS, Holmes MC, Zhang L, Gregory PD, Rebar EJ (2011) A TALE nuclease architecture for efficient genome editing. Nat Biotechnol 29(2):143–148. doi:10.1038/nbt.1755
Moehle EA, Rock JM, Lee YL, Jouvenot Y, DeKelver RC, Gregory PD, Urnov FD, Holmes MC (2007) Targeted gene addition into a specified location in the human genome using designed zinc finger nucleases. Proc Natl Acad Sci USA 104(9):3055–3060. doi:10.1073/pnas.0611478104
Moscou MJ, Bogdanove AJ (2009) A simple cipher governs DNA recognition by TAL effectors. Science 326(5959):1501. doi:10.1126/science.1178817
Orlando SJ, Santiago Y, DeKelver RC, Freyvert Y, Boydston EA, Moehle EA, Choi VM, Gopalan SM, Lou JF, Li J, Miller JC, Holmes MC, Gregory PD, Urnov FD, Cost GJ (2010) Zinc-finger nuclease-driven targeted integration into mammalian genomes using donors with limited chromosomal homology. Nucleic Acids Res 38(15):e152. doi:10.1093/nar/gkq512
Orlowski J, Boniecki M, Bujnicki JM (2007) I-Ssp6803I: the first homing endonuclease from the PD-(D/E)XK superfamily exhibits an unusual mode of DNA recognition. Bioinformatics 23(5):527–530. doi:10.1093/bioinformatics/btm007
Pabo CO, Peisach E, Grant RA (2001) Design and selection of novel Cys2His2 zinc finger proteins. Annu Rev Biochem 70:313–340. doi:10.1146/annurev.biochem.70.1.313
Palgrave CJ, Gilmour L, Lowden CS, Lillico SG, Mellencamp MA, Whitelaw CB (2011) Species-specific variation in RELA underlies differences in NF-κB activity: a potential role in African swine fever pathogenesis. J Virol 85(12):6008–6014. doi:10.1128/JVI.00331-11
Pavletich NP, Pabo CO (1991) Zinc finger-DNA recognition: crystal structure of a Zif268-DNA complex at 2.1 A. Science 252(5007):809–817
Petersen B, Lucas-Hahn A, Oropeza M, Hornen N, Lemme E, Hassel P, Queisser AL, Niemann H (2008) Development and validation of a highly efficient protocol of porcine somatic cloning using preovulatory embryo transfer in peripubertal gilts. Cloning Stem Cells 10(3):355–362. doi:10.1089/clo.2008.0026
Posfai G, Kolisnychenko V, Bereczki Z, Blattner FR (1999) Markerless gene replacement in Escherichia coli stimulated by a double-strand break in the chromosome. Nucleic Acids Res 27(22):4409–4415
Proudfoot C, Carlson DF, Huddart R, Long CR, Pryor JH, King TJ, Lillico SG, Mileham AJ, McLaren DG, Whitelaw CB, Fahrenkrug SC (2015) Genome edited sheep and cattle. Transgenic Res 24(1):147–153. doi:10.1007/s11248-014-9832-x
Puchta H (1999) Use of I-Sce I to induce DNA double-strand breaks in Nicotiana. Methods Mol Biol (Clifton, NJ) 113:447–451. doi:10.1385/1-59259-675-4:447
Puchta H (2002) Gene replacement by homologous recombination in plants. Plant Mol Biol 48(1–2):173–182
Radding CM (1982) Homologous pairing and strand exchange in genetic recombination. Annu Rev Genet 16:405–437. doi:10.1146/annurev.ge.16.120182.002201
Renfer E, Amon-Hassenzahl A, Steinmetz PR, Technau U (2010) A muscle-specific transgenic reporter line of the sea anemone, Nematostella vectensis. Proc Natl Acad Sci USA 107(1):104–108. doi:10.1073/pnas.0909148107
Rogers CS, Stoltz DA, Meyerholz DK, Ostedgaard LS, Rokhlina T, Taft PJ, Rogan MP, Pezzulo AA, Karp PH, Itani OA, Kabel AC, Wohlford-Lenane CL, Davis GJ, Hanfland RA, Smith TL, Samuel M, Wax D, Murphy CN, Rieke A, Whitworth K, Uc A, Starner TD, Brogden KA, Shilyansky J, McCray PB Jr, Zabner J, Prather RS, Welsh MJ (2008) Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science 321(5897):1837–1841. doi:10.1126/science.1163600
Rosen LE, Morrison HA, Masri S, Brown MJ, Springstubb B, Sussman D, Stoddard BL, Seligman LM (2006) Homing endonuclease I-CreI derivatives with novel DNA target specificities. Nucleic Acids Res 34(17):4791–4800. doi:10.1093/nar/gkl645
Rouet P, Smih F, Jasin M (1994a) Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc Natl Acad Sci USA 91(13):6064–6068
Rouet P, Smih F, Jasin M (1994b) Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol Cell Biol 14(12):8096–8106
Sander JD, Cade L, Khayter C, Reyon D, Peterson RT, Joung JK, Yeh JR (2011) Targeted gene disruption in somatic zebrafish cells using engineered TALENs. Nat Biotechnol 29(8):697–698. doi:10.1038/nbt.1934
Seligman LM, Chisholm KM, Chevalier BS, Chadsey MS, Edwards ST, Savage JH, Veillet AL (2002) Mutations altering the cleavage specificity of a homing endonuclease. Nucleic Acids Res 30(17):3870–3879
Shen B, Zhang W, Zhang J, Zhou J, Wang J, Chen L, Wang L, Hodgkins A, Iyer V, Huang X, Skarnes WC (2014) Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat Methods 11(4):399–402. doi:10.1038/nmeth.2857
Siebert R, Puchta H (2002) Efficient repair of genomic double-strand breaks by homologous recombination between directly repeated sequences in the plant genome. Plant Cell 14(5):1121–1131
Silva G, Poirot L, Galetto R, Smith J, Montoya G, Duchateau P, Paques F (2011) Meganucleases and other tools for targeted genome engineering: perspectives and challenges for gene therapy. Curr Gene Ther 11(1):11–27
Smih F, Rouet P, Romanienko PJ, Jasin M (1995) Double-strand breaks at the target locus stimulate gene targeting in embryonic stem cells. Nucleic Acids Res 23(24):5012–5019
Smith J, Bibikova M, Whitby FG, Reddy AR, Chandrasegaran S, Carroll D (2000) Requirements for double-strand cleavage by chimeric restriction enzymes with zinc finger DNA-recognition domains. Nucleic Acids Res 28(17):3361–3369
Smith J, Grizot S, Arnould S, Duclert A, Epinat JC, Chames P, Prieto J, Redondo P, Blanco FJ, Bravo J, Montoya G, Paques F, Duchateau P (2006) A combinatorial approach to create artificial homing endonucleases cleaving chosen sequences. Nucleic Acids Res 34(22):e149. doi:10.1093/nar/gkl720
Sussman D, Chadsey M, Fauce S, Engel A, Bruett A, Monnat R Jr, Stoddard BL, Seligman LM (2004) Isolation and characterization of new homing endonuclease specificities at individual target site positions. J Mol Biol 342(1):31–41. doi:10.1016/j.jmb.2004.07.031
Szczepek M, Brondani V, Buchel J, Serrano L, Segal DJ, Cathomen T (2007) Structure-based redesign of the dimerization interface reduces the toxicity of zinc-finger nucleases. Nat Biotechnol 25(7):786–793. doi:10.1038/nbt1317
Takeuchi H, Georgiev O, Fetchko M, Kappeler M, Schaffner W, Egli D (2007) In vivo construction of transgenes in Drosophila. Genetics 175(4):2019–2028. doi:10.1534/genetics.106.065920
Tan W, Carlson DF, Lancto CA, Garbe JR, Webster DA, Hackett PB, Fahrenkrug SC (2013) Efficient nonmeiotic allele introgression in livestock using custom endonucleases. Proc Natl Acad Sci USA 110(41):16526–16531. doi:10.1073/pnas.1310478110
Terns MP, Terns RM (2011) CRISPR-based adaptive immune systems. Curr Opin Microbiol 14(3):321–327. doi:10.1016/j.mib.2011.03.005
Tesson L, Usal C, Menoret S, Leung E, Niles BJ, Remy S, Santiago Y, Vincent AI, Meng X, Zhang L, Gregory PD, Anegon I, Cost GJ (2011) Knockout rats generated by embryo microinjection of TALENs. Nat Biotechnol 29(8):695–696. doi:10.1038/nbt.1940
Tsai SQ, Wyvekens N, Khayter C, Foden JA, Thapar V, Reyon D, Goodwin MJ, Aryee MJ, Joung JK (2014) Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol 32(6):569–576. doi:10.1038/nbt.2908
Urnov FD, Miller JC, Lee YL, Beausejour CM, Rock JM, Augustus S, Jamieson AC, Porteus MH, Gregory PD, Holmes MC (2005) Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 435(7042):646–651. doi:10.1038/nature03556
van den Heuvel M, Sorop O, Koopmans SJ, Dekker R, de Vries R, van Beusekom HM, Eringa EC, Duncker DJ, Danser AH, van der Giessen WJ (2012) Coronary microvascular dysfunction in a porcine model of early atherosclerosis and diabetes. Am J Physiol Heart Circ Physiol 302(1):H85–H94. doi:10.1152/ajpheart.00311.2011
Vasquez KM, Marburger K, Intody Z, Wilson JH (2001) Manipulating the mammalian genome by homologous recombination. Proc Natl Acad Sci USA 98(15):8403–8410. doi:10.1073/pnas.111009698
Voytas DF, Joung JK (2009) Plant science. DNA binding made easy. Science 326(5959):1491–1492. doi:10.1126/science.1183604
Wang J, Friedman G, Doyon Y, Wang NS, Li CJ, Miller JC, Hua KL, Yan JJ, Babiarz JE, Gregory PD, Holmes MC (2012) Targeted gene addition to a predetermined site in the human genome using a ZFN-based nicking enzyme. Genome Res 22(7):1316–1326. doi:10.1101/gr.122879.111
Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R (2013) One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153(4):910–918. doi:10.1016/j.cell.2013.04.025
Watanabe M, Umeyama K, Matsunari H, Takayanagi S, Haruyama E, Nakano K, Fujiwara T, Ikezawa Y, Nakauchi H, Nagashima H (2010) Knockout of exogenous EGFP gene in porcine somatic cells using zinc-finger nucleases. Biochem Biophys Res Commun 402(1):14–18
Watanabe M, Nakano K, Matsunari H, Matsuda T, Maehara M, Kanai T, Kobayashi M, Matsumura Y, Sakai R, Kuramoto M, Hayashida G, Asano Y, Takayanagi S, Arai Y, Umeyama K, Nagaya M, Hanazono Y, Nagashima H (2013) Generation of interleukin-2 receptor gamma gene knockout pigs from somatic cells genetically modified by zinc finger nuclease-encoding mRNA. PLoS ONE 8(10):e76478. doi:10.1371/journal.pone.0076478
Whitworth KM, Lee K, Benne JA, Beaton BP, Spate LD, Murphy SL, Samuel MS, Mao J, O’Gorman C, Walters EM, Murphy CN, Driver J, Mileham A, McLaren D, Wells KD, Prather RS (2014a) Use of the CRISPR/Cas9 system to produce genetically engineered pigs from in vitro-derived oocytes and embryos. Biol Reprod 91(3):78. doi:10.1095/biolreprod.114.121723
Whitworth KM, Lee K, Benne JA, Beaton BP, Spate LD, Murphy SL, Samuel MS, Mao J, O’Gorman C, Walters EM, Murphy CN, Driver JP, Mileham A, McLaren D, Wells KD, Prather RS (2014b) Use of the CRISPR/Cas9 system to produce genetically engineered pigs from in vitro-derived oocytes and embryos. Biol Reprod. doi:10.1095/biolreprod.114.121723
Whyte JJ, Prather RS (2011) Zinc finger nucleases to create custom-designed modifications in the swine (Sus scrofa) genome. J Anim Sci. doi:10.2527/jas.2011-4546
Whyte JJ, Zhao J, Wells KD, Samuel MS, Whitworth KM, Walters EM, Laughlin MH, Prather RS (2011) Gene targeting with zinc finger nucleases to produce cloned eGFP knockout pigs. Mol Reprod Dev 78(1):2. doi:10.1002/mrd.21271
Wiedenheft B, Sternberg SH, Doudna JA (2012) RNA-guided genetic silencing systems in bacteria and archaea. Nature 482(7385):331–338. doi:10.1038/nature10886
Windbichler N, Papathanos PA, Catteruccia F, Ranson H, Burt A, Crisanti A (2007) Homing endonuclease mediated gene targeting in Anopheles gambiae cells and embryos. Nucleic Acids Res 35(17):5922–5933. doi:10.1093/nar/gkm632
Yang D, Yang H, Li W, Zhao B, Ouyang Z, Liu Z, Zhao Y, Fan N, Song J, Tian J, Li F, Zhang J, Chang L, Pei D, Chen YE, Lai L (2011) Generation of PPARgamma mono-allelic knockout pigs via zinc-finger nucleases and nuclear transfer cloning. Cell Res. doi:10.1038/cr.2011.70
Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R (2013) One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 154(6):1370–1379. doi:10.1016/j.cell.2013.08.022
Yu BJ, Kang KH, Lee JH, Sung BH, Kim MS, Kim SC (2008) Rapid and efficient construction of markerless deletions in the Escherichia coli genome. Nucleic Acids Res 36(14):e84. doi:10.1093/nar/gkn359
Yu S, Luo J, Song Z, Ding F, Dai Y, Li N (2011) Highly efficient modification of beta-lactoglobulin (BLG) gene via zinc-finger nucleases in cattle. Cell Res 21(11):1638–1640. doi:10.1038/cr.2011.153
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Petersen, B., Niemann, H. Molecular scissors and their application in genetically modified farm animals. Transgenic Res 24, 381–396 (2015). https://doi.org/10.1007/s11248-015-9862-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11248-015-9862-z