
Abstract
In an attempt to explore the method of nitrohydroxylation which is fairly underdeveloped, our tridentate NHC–amidate–alkoxide containing palladium catalyst was used to nitrohydroxylate a variety of olefins in the presence of nitric acid. These reactions furnished β-nitro alcohols selectively to serve as a direct method for the synthesis of such compounds from various kinds of olefins. Dioxane served as an effective solvent, particularly in conjunction with TFA and AgNO3, which selectively generated the desired products. Vinyl arenes and other olefins produced a wide range of desired products in moderate to good yields.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Development of an effectual method for synthesizing poly-functional small molecules as building blocks is indispensable in a number of applications such as biological studies, new material discovery, and molecular synthesis. One such family of small molecules with great synthetic importance is nitro alcohols and their derivatives. Due to their simplistic derivatization of numerous compounds, nitro alcohols have great significance for organic chemistry research as well as industrial feedstocks. Bordwell devised nitroacetoxylation by adopting acetyl nitrate with styrene to afford a mixture of nitroalkenes, β-nitro nitrates, and β-nitro acetates [1,2,3,4]. Even though, Bordwell’s methodology offered simultaneous C–O and C–N bond formation, it was still deficient to be a useful method due to unwieldly procedure and conditions. Furthermore, poor chemoselectivity and diastereoselectivity were observed as a problem as well. With extensive literature search, direct nitro-hydroxylation was found to be underdeveloped and underutilized due to low efficiency, poor chemoselectivity and diastereoselectivity, and drastic conditions.
Successful nitrohydroxylation would synthesize β-nitro alcohols (or nitro aldols) [5], which can also be prepared by the Henry reaction [6]. While the Henry reaction utilizes carbonyl and nitroalkane substrates, nitrohydroxylation utilizes alkenes and nitric acid, both of which are readily available. Though, the Henry reaction has exhibited very few successful cases with ketones, generating disubstituted nitro alcohols [7, 8]. The direct regioselective addition of nitric acid across an olefin through nitrohydroxylation could not only provide an efficient and straightforward manner for generating nitro alcohols but also furnish excellent alternative to the Henry reaction with wider scope of reactivity and applicability. Nevertheless, most reports on the use of nitric acid in conjunction with alkenes have focused on the synthesis of nitro olefins [9]. Herein, we report an effective method for synthesizing β-nitro alcohols from olefins and nitric acid using our tridentate NHC–amidate–alkoxide containing palladium catalyst 1 (Fig. 1).

Palladium catalyst of tridentate NHC–amidate–alkoxide ligands [11]
2 Experimental
2.1 Solvent and Temperature Variation
To an oven dried screw cap vial equipped with a stir bar and the threads covered with Teflon tape, were added catalyst 1 (0.05 mmol), AgNO3 (0.1 mmol), and desired solvent (1.5 mL). After the reaction mixture was stirred for 10 min at room temperature to activate the catalyst, styrene (1.0 mmol) and nitric acid (2.0 mmol) were added. The reaction was then capped and stirred for 24 h at desired temperatures (50–150 °C). The crude reaction mixture was filtered through a pad of Celite and then subjected to flash column chromatography using a polarity gradient system (hexanes to 2:1 hexanes/ethyl acetate) to obtain the desired product.
2.2 Catalyst Comparison
Desired catalyst (0.05 mmol) was added to 1.5 mL dioxane in pre-dried screw cap vial equipped with a stir bar. Styrene (1.0 mmol) and nitric acid (2.0 mmol) were added to the resulting solution. The reaction was then capped and stirred for 24 h at 90 °C. The crude reaction mixture was filtered through a pad of Celite and then subjected to flash column chromatography using a polarity gradient system (hexanes to 2:1 hexanes/ethyl acetate) to obtain the desired product.
2.3 General Nitrohydroxylation
Catalyst 1a or 1b (0.05 mmol) and AgNO3 (0.1 mmol) were added into pre-dried screw cap vial equipped with 1.5 mL dioxane. The reaction mixture was stirred for 10 min at room temperature to activate the catalyst. To the resulting solution, were added olefin (1.0 mmol) and nitric acid (2.0 mmol). The reaction was then capped and stirred for 24 h at 90 °C. The crude reaction mixture was filtered through a pad of Celite and then subjected to flash column chromatography using a polarity gradient system (hexanes to 2:1 hexanes/ethyl acetate) to obtain the desired product.
3 Results and Discussion
While we were exploring the synthesis of β-nitro alcohols with palladium catalyst 1a, we found direct nitrohydroxylation on styrene in the presence of nitric acid (Scheme 1). Initially, this reaction was run by using styrene as a substrate and two equivalents of nitric acid in acetonitrile at 50 °C for 20 h. In this reaction we were able to observe nitrohydroxylation to form β-nitro alcohols 2 (28% yield based on styrene) as well as about 3% of dinitro compound 3.

Nitrohydroxylation of styrene by catalyst
With this result, we tried to investigate direct nitrohydroxylation on olefin. As the first step, solvent variation was monitored (Table 1, entries 1–5). In DCM, nitro alcohol 2 was obtained in 31% (entry 1). While lower conversion yields (15 and 28%, respectively) were observed in DMF and acetonitrile (entries 2 and 3), β-nitro alcohol 2 was produced with higher yields (38 and 41%) repeatedly in ether solvents including THF and dioxane (entries 4 and 5). In addition, since dioxane has a higher boiling point than THF, we were able to raise the reaction temperature to 90 °C and detected a higher yield of 55% (entry 6). However, it produced a lower yield when the reaction temperature was further increased to 150 °C (entry 7). This might be due to the decomposition of the catalyst. For the selectivity between 2 and 3, acetonitrile produced 2 with high selectivity (91%), however other solvents showed 75–80% selectivity for 2.
With the optimal choice of solvent conditions (Table 1, entry 6), various commercially available catalysts were directly compared with catalyst 1a and the results are presented in Table 2.
Without catalyst (entry 1), we didn’t detect nitrohydroxylation at all. Both nickel sources (entries 2 and 3) showed relatively low yields (19 and 18%, respectively). Palladium catalysts were relatively efficient. In the case of PdCl2 (entry 4) and Pd(PPh3)2 (entry 5), nitro alcohol 2 was produced in 22%. Pd(CH3CN)2Cl2 and Pd(OAc)2 gave slightly higher yields (26%, entries 6 and 7). These results showed nitrohydroxylation could be performed with various commercially available catalysts but in lower yields than 1a.
Pursuing optimal conditions, we investigated the use of additives, and found the addition of an acid was beneficial. Trifluoroacetic acid was previously used as a solvent for nitration reactions, and was known to prevent the formation of dinitrogen pentoxide, N2O5, which would hamper nitrohydroxylation [10]. As shown in Scheme 2, two equivalents of TFA were added to our conditions, improving the yields of nitro alcohols, i.e., from 55 to 65% for 2. For the possible enantioselective catalysis due to the stereogenic α position, we employed the chiral palladium catalyst 1b, which furnished high enantioselectivities for boron Heck-type reaction [11, 12]. This chiral catalyst 1b showed similar results to achiral catalyst 1a in terms of yield for 2 but it produced a racemic mixture. With these conditions in hand, we examined the substrate scope of the reaction with 1b (vide infra) while hoping for possible asymmetric catalysis, which didn’t occur.

Nitrohydroxylation of vinyl arenes with nitric acid. All the products were isolated and the yields were based on isolation
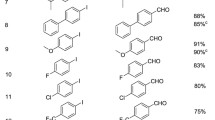
First, attention was focused on the conversion of vinyl arenes (Scheme 2). Alkyl substituted vinyl arenes furnished the desired nitro alcohols 4 and 5 in good yields of 54 and 64%, respectively. Vinyl naphthalene gave the desired product 6 in a modest 47% yield. The addition of a strong electron donating group to the arene drastically decreased the yield, as demonstrated by the use of 4-methoxy styrene to give the product 7 in a low yield of 24%. This might be due to the polymerization of 4-methoxy styrene under acidic conditions, which was observed in our previous work, hydroalkenylation of arenes [13]. Halogen substituted vinyl arenes produced the desired nitro alcohols 8, 9, 10, and 11 in moderate to low yields. However, it is noteworthy that these halogen substituted vinyl arenes were compatible with desired chemoselectivity and no Heck or homo coupling products were observed. This feature should lend these reactions to use in tandem or one-pot multi-component dual reaction sequences.
In addition to vinyl arenes, several other alkenes were found to be viable substrates for nitrohydroxylation using this methodology (Scheme 3). The relatively unactivated olefin 4-phenyl-1-butene was converted to the desired product 12 in a 51% yield and allyl benzyl ether gave the desired nitro alcohol 13 at 52%. In the case of α,β-unsaturated carbonyl compounds, ethyl vinyl ketone reacted modestly to give 14 in a 46% yield, while tert-butyl acrylate which has an electron donating group reacted poorly to give 15 in only 29% yield.

Nitrohydroxylation of unconjugated olefins with nitric acid. All the products were isolated and the yields were based on isolation
Impressively, it was found that this reaction was compatible to geminal disubstituted olefins, yielding nitro-alcohols possessing a tertiary alcohol (Scheme 4). The nitro alcohol 16 was generated in poor yield, though cleanly, from tert-butyl methacrylate. α-Methyl styrene was converted to the desired compound 17 in a good yield of 70%. The yields of 16 and 17 from disubstituted olefins showed similar yields to those of 2 and 15 from mono-substituted olefins, respectively. The alkyl disubstituted olefins such as 2-ethylbutene gave the corresponding desired product 18 in 53% yield.

Nitrohydroxylation of geminal disubstituted olefins with nitric acid. All of product was isolated to calculate yield
The feasibility of using this methodology on vicinal disubstituted olefins was also tested. While substrates tested thus far were terminal and gave no possibility for syn and anti isomers, we knew that this problem could arise for using vicinally disubstituted olefins. The use of both β-methyl styrene and indene unfortunately resulted in the formation of both the syn and anti addition products. In the case of β-methyl styrene, a 1 to 1 mixture of both isomers 19 was observed while indene showed a preference for the anti isomer 20 in a 2 to 1 ratio (Scheme 5).

Nitrohydroxylation vicinal disubstituted olefins with nitric acid. All the products were isolated and the yields were based on isolation
Lack of possible enantioselectivity and poor diastereoselectivity led us to considering the stepwise addition of the nitro group and hydroxyl to an alkene as proposed in Scheme 6. Under our conditions, we believe nitronium ion (23) would form slowly by the aid of a Pd(II) catalyst, so the potential side reactions such as aromatic nitration, dinitration, nitroso nitrate formation would be minimized. Then, the Pd–π complex 24 would undergo nitration with the external nitronium ion, in which both syn and anti products can be generated [14]. The resulting nitration intermediate 25 would lead to the nitrohydroxylation product such as 2 by reductive elimination. In a cyclic system such as indene, the nitro group would approach in the anti position to the Pd moiety to afford the anti isomer as the major in 20 while the acyclic system would give both syn and anti in the same amounts (19). Underway are our efforts to make the addition of NO2 and OH groups in a concerted manner to offer higher selectivities.

Proposed mechanism and reaction pathway
Due to numerous uses of nitro alcohols and their derivatives, palladium catalyzed nitrohydroxylation of olefins is undoubtedly a field of research which needs to be explored in greater detail. Previously reported methods were often limited to narrow substrate scope under harsh and long reaction conditions with the use of large excess nitrate sources. In contrast, the conversion of various olefins to corresponding β-nitro alcohols with nitric acid was accomplished via the use of novel tridentate NHC–amidate–alkoxide containing catalysts. Our reaction turned out to be an efficient way of utilizing many different vinyl arenes under mild conditions with short reaction time in moderate to good yields. Moreover, these reactions occurred with superior regio- and chemo-selectivities. It should provide a straightforward and practical method for generating such nitro alcohols which can be employed in synthetic organic, bioorganic, and material chemistry. Our finding demonstrates that nitrohydroxylation has a potential to serve as an efficient alternative for amino alcohol synthesis. However, this direct synthesis of β-amino alcohols by nitrohydroxylation has not generated enantioselectivity. Therefore, further research is being directed to produce high enantioselectivity through the modification of chiral catalysts 1b or the application of known chiral catalysts.
References
Bordwell FG, Garbisch EW (1960) J Chem Soc 82(14):3588–3598
Bordwell FG, Garbisch EW (1963) J Org Chem 28(7):1765–1769
Bordwell FG, Garbisch EW (1962) J Org Chem 27(9):3049–3055
Bordwell FG, Biranowski JB (1967) J Org Chem 32(3):629–634
Ono N (2001) The nitro group in organic synthesis. Chapter 3. Wiley-VCH, Weinheim
Bodkin JA, Mcleod MD (2002) J Chem Soc 1:2733–2746
Palomo C, Oiarbide M, Laso A (2007) Eur J Org Chem 2007(16):2561–2574
Luzzio FA (2001) Tetrahedron 57(6):915–945
Thimmaiah G, Narayanaswamy N (March 2, 2017) Compounds as Stimuli-responsive Probes, Methods and Applications thereof. WO 2017033163
Shen G, Zhao L, Liu W, Huang X, Song H, Zhang T (2017) Synth Commun 47(1):10–14
Sakaguchi S, Yoo KS, O’Neill J, Jung KW (2008) Angew Chem Int Ed 47:9326–9329
Yoo KS, O’Neill J, Sakaguchi S, Giles R, Lee JH, Jung KW (2009) J Org Chem 75:95–101
Zargari N, Gilles R, Kim Y, Kaneshiro K, Runburg R, Park J, LaCroix K, Narain R, Lee BD, Lee JH, Jung KW (2016) Tetrahedron Lett 57:815–818
Esteves PM, Carneiro JWM, Cardoso SP, Barbosa AGH, Laali KK, Rasul G, Prakash GKS, Olah GA (2003) J Am Chem Soc 125(16):4836–4849
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
We do not have any potential conflicts of interest.
Research Involving Human Participants and/or Animals
This work doesn’t involve any human participants or animals.
Informed Consent
We do not have any informed consent.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
O’Neill, J., Lee, J.H., Kim, S. et al. Nitrohydroxylation of Olefins with Nitric Acid Using Tridentate NHC–Amidate–Alkoxide Containing Palladium Catalysts. Top Catal 61, 630–635 (2018). https://doi.org/10.1007/s11244-018-0915-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-018-0915-4