
Abstract
The binding of molecules to the surface of nanoparticles (NPs) for the use as ligands to manipulate the catalytic properties of NPs is an emerging research area. Various studies with interesting results have been reported in the past few years, but it seems not clear how these findings could be merged into some kind of unified picture, describing the mechanism of action of ligands in heterogeneous catalysis. The aim of this article is to summarize some of the recent achievements in this field with focus on discussing these results using concepts from heterogeneous and homogeneous catalysis. By this it is attempted to separate the influence of ligands into (i) changing the surface properties and (ii) acting as a function above or perpendicular to the surface. The first aspect can be rationalized by the knowledge from bimetallic catalysis. In contrast, the second proposes the relevance of ligand–reactant interactions, as known from homogeneous catalysis, in order to manipulate adsorption, activation, and conversion of reactants. As the application of ligands in heterogeneous catalysis is still a young research field and the full potential of the approach still unknown, this article does not claim to give a complete summery of all results gained within this field. Instead, the author aims to present a picture that may give some guidance for future studies in this area, based on established knowledge from homo- and heterogeneous catalysis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
A typical metalorganic homogeneous catalyst is a metal complex formed from a single metal atom or an ion, surrounded by ligands that bind to the metal center. If free coordination sites are available at the metal center, a reactant can be adsorbed, activated, and catalytically converted. For homogeneous catalysts ligands are prerequisite because they mediate the solubility of the active metal complex in the reaction medium and protect the metal center from deactivation via e.g. sintering or precipitation. The heterogeneous analogue to a metalorganic compound is a supported nanoparticle. When considering ligands merely as stabilizers for the catalytic metal it becomes apparent that ligands are not required for a heterogeneous catalyst, because the particles are already stabilized by the support. Furthermore, the binding of ligands onto the surface of a nanoparticle leads to the formation of self-assembled monolayers that may eventually block the catalytic surface. Therefore, it is usually concluded that ligands reduce the catalytic activity of a heterogeneous catalyst [1], which at first sight is an undesired effect.
When taking a deeper look into the role of ligands in homogeneous catalysis it becomes clear that ligands exhibit abilities beyond their use as stabilizers that may also be of potential interest for heterogeneous catalysis. Ligands can electronically modify the metal center to which they bind [2]. This alters its reactivity which in turn can lead to changes in activity and selectivity. With respect to their structure, ligands can sterically and electronically interact with reactants, which enables for a unique control over selectivity [3]. Moreover, they exhibit the ability to open alternative reaction pathways with enhanced rates compared to the purely metal catalyzed reaction, which is related to as ligand acceleration [4].
As highlighted in several guidelines for 21st century catalysis research the understanding and developing of new strategies to control selectivity, especially in heterogeneous catalysis, will be one of the major research tasks for the next decades [5–7]. This requires manipulation of essential material properties. Typical strategies for supported nanoparticles are tuning the particle size [8, 9], using a second metal (bimetallic nanoparticles) [10, 11], or the creation of new active sites by e.g. inducing strong-metal support interactions (SMSI) [12]. The basic idea of these approaches is to manipulate the surface properties and by this the adsorption and activation of the reactants on the surface. As discussed later in more detail, ligands can be applied in a similar way to alter the surface properties of particles. As the selectivity of a heterogeneous catalyst can be tuned in this way [13], it can be concluded that ligands serve as a novel approach for tuning selectivity. The potential of ligands for heterogeneous catalysis has not merely been identified by academic research but also by chemical industry. E.g. the BASF developed a commercially available heterogeneous catalysts that consists of supported Pd NPs, being covered with ligands [14]. This catalyst is highly selective for the semi-hydrogenation of alkynes to alkenes, serving as a lead-free and therefore green alternative to the conventional Lindlar catalyst.
As indicated above, one ability to control selectivity in homogeneous catalysis is to achieve specific ligand–reactant interactions [15, 16]. By this the activation of the reactant can be altered or even alternative reaction pathways that are controlled by the ligand may become feasible enabling for a unique control of selectivity [4]. Considering the impressive progress that has been achieved in this way within the field of metalorganic catalysis raises the question if it is feasible to use ligand–reactant interactions for the control of selectivity in heterogeneous catalysis. Two approaches can be found that have been explored in the past and evidence that ligands may indeed be suitable for this task:
-
1.
Use of dispersed colloidal nanoparticles applied as “quasi-homogeneous” catalysts.
-
2.
Application of chiral modifiers for supported nanoparticle catalysts.
In the 1980s and 1990s considerable progress has been achieved within the field of colloidal nanoparticles formed from metals being of catalytic importance like Pd, Pt, or Ru [17]. Colloidal nanoparticles are synthesized in solution via reduction of a metal precursor. A stabilizer is usually required in order to shield the particle surface, which suppresses particle sintering. Reduction can be achieved by solvents such as alcohols [18, 19], using strong reducing agents like NaBH4 [20], and furthermore by electrochemical methods [21]. Most commonly applied stabilizers are polymers such as polyvinylpyrrolidone (PVP) [22], standard alkyl surfactants [17], but also ligands that are known from homogeneous catalysis are suitable for stabilizing dispersed colloidal nanoparticles [23, 24]. The stabilizers bind coordinatively or covalently to the particle surface similar as ligands to a metal center. In this way the particle’s surface energy is lowered and a protecting shell is formed that keeps the particles at distance to inhibit sintering that would cause fade of the nanostructure. Colloidal syntheses can be applied for the preparation of heterogeneous model catalysts, because the resulting particles can be deposited onto any given support material and the protecting agents be removed by thermal treatments or using ozone [25–27]. In this way the influence of particle size [28], shape [29], support [20] and in the case of bimetallic materials the effect of the chemical composition [30] on the catalytic properties can be studied representatively and in a systematic manner. However, when maintaining the stabilizing shell, colloidal nanoparticles can also be applied as “quasi-homogeneous” catalysts, meaning dispersed in the reaction medium like a metal complex [31]. The application of colloidal nanoparticles, stabilized by specific ligands from metalorganic chemistry as “quasi-homogeneous” catalysts revealed that selectivities can be altered by the presence of ligands [32, 33]. Furthermore, even specific selectivities like stereoselectivity could be achieved [34]. In order to establish stereoselectivities it is prerequisite to achieve an asymmetry bias. In homogeneous catalysis the ligand is the chiral element of the catalyst and the ligand–reactant interaction is considered as the source for the asymmetric bias. The fact that stereoselective catalytic reactions can be performed with ligand-stabilized colloidal nanoparticles hence indicates that ligand–reactant interaction can also be established on particle surfaces and used to control the selectivity of catalytic nanoparticles.
The approach of using colloidal nanoparticles as catalysts has apparently its origin in homogeneous catalysis. In contrast, the application of so-called chiral modifiers was introduced by groups from the field of heterogeneous catalysis. A chiral modifier is an organic molecule that prior to starting a catalytic conversion with a supported nanoparticle catalyst is dissolved together with the reactant in the reaction medium [35]. Depending on its binding strength to the particle surface and its solvation energy the modifier is in an adsorption–desorption equilibrium, which determines the ratio of unmodified to modified sites of the catalytic surface [36, 37]. Whereas over unmodified sites a racemic reaction occurs, the reactant can be stereoselectively converted when interacting appropriately with an adsorbed modifier [38]. The most investigated modifiers are cinchona alkaloids (see Fig. 1). The aromatic moiety (highlighted with green) acts as the anchoring group for the modifier to bind to the particle surface. Only when this part of the modifier adsorbs flat on the surface, high stereoselectivities can be achieved [39]. The red highlighted part of the modifier contains the stereogenic center with an amino group that has identified to be essential for the asymmetric induction, as e.g. methylation of this group leads to complete loss of stereoselectivity [40]. The origin of chiral induction has been intensively investigated and discussed in the past two decades [38]. Today it is generally accepted that it originates from a one to one interaction of an adsorbed modifier with an adsorbed reactant. The working principle of a modifier seems hence to be quite similar to ligands in homogeneous catalysis. The fact that the modifier is soluble in the reaction medium complicates its applicability as a serve bound species. If the modifier surface coverage is too low, the catalyst exhibits free metal surface over which a purely metal-catalyzed and thus a racemic reaction proceeds. If, in contrast, the coverage is too high, the adsorption mode shifts from a flat-lying modifier to a tilted mode, which is not suitable to form the desired modifier–reactant interaction [41]. Another issue, beside any surface coverage dependent changes in the catalytic properties, is that the aromatic moiety undergoes hydrogenation under catalytic conditions [42]. The modifier structure is hence altered and its adsorption strength on the particle surface lowered which as a result causes losses of stereoselectivity [43].

Cinchona alkaloids have intensively been studied as chiral modifiers for supported nanoparticle catalysts with significant success in terms of stereoselectivity for several reactants. The green marked part of the modifier serves as the anchoring group to adsorb on the particle surface, whereas the red part is responsible for the asymmetric bias. A detailed description of the underlying mechanistic details can be found in the literature [38]
In order to overcome the problems that are accompanied with the use of a molecule as a modifier, the following strategies could be followed:
-
1.
Use of molecules that are not reactive under applied reaction conditions.
-
2.
Fixation of the modifier on the surface in order to suppress the possibility of an adsorption–desorption equilibrium.
As above mentioned the strength of adsorption of a molecule is determined by its binding strength to the catalytic surface and its solubility in the reaction medium. Therefore, fixation can be achieved by increasing the binding strength (e.g. the use of sulfur-based anchoring group) [44, 45] and by using a reaction medium in which the molecule that is supposed to be bound to the surface is not soluble [46]. When working with gaseous reactants there are of course no solubility related issues that have to be taken into consideration. Instead it is merely important to keep the reaction temperature low enough to avoid desorption of the desired surface bound species.
Based on the above discussion it is reasonable to make a clear distinction between the terms “modifier” and “ligand” to avoid any confusion. A “modifier” is a molecule that is soluble in the reaction medium and has thus to be considered as being in an adsorption–desorption equilibrium under reaction conditions [40]. In contrast, a “ligand” is a molecule that is kept fixated on the surface under catalytic conditions [1].
As already indicated above, the use of modifiers for supported nanoparticle catalysts in liquid media is an established approach for the control of stereoselectivity. Significant progress has been achieved within this field and reviews summarizing the most relevant aspects have been published by different groups [38, 47, 48]. The idea of fixating a molecule to the surface of the catalyst in order to apply it as a ligand has however just recently started to attract attention [1] and it is yet not clear what potential this approach bears.
The aim of this article is to summarize some of the recent work on the application of ligands in heterogeneous catalysis and to provide a discussion on how the influence of ligands on the catalytic properties could be rationalized. For the latter, concepts from both, heterogeneous and homogeneous catalysis, are discussed regarding their validity as models in order to explain experimental observations. The basic idea is to understand the ligand as a surface bound species that can:
-
1.
Alter the electronic and geometric surface properties,
-
2.
Sterically and electronically interact with the reactant.
The first aspect clearly represents a conceptual way of thinking from heterogeneous catalysis and focusses only on what occurs on the surface (two dimensional catalyst modification). In contrast, the second aspect is a common strategy in homogenous catalysis to manipulate the catalytic properties and suggests that a ligand can also act as function above or perpendicular to the surface for manipulating the reactant activation. This basic idea of separating the influence of ligands in this way is illustrated in Fig. 2.

The presented idea to rationalize the influence of ligands on the catalytic properties of supported nanoparticles is to separate it into (i) changes of surface properties (red label and arrows) and (ii) ligand–reactant interactions (blue label and arrow). As the latter take place above the surface, ligands are proposed to be considered as a selectivity controlling function perpendicular to the surface in case appropriate ligand–reactant interactions can be established
In the following examples will be discussed that evidence the validity of both these conceptual ways of thinking. Thereby, it is focused on the control of chemoselectivity and furthermore stereoselectivity as two of the greatest challenges in heterogeneous catalysis. By this it is tried to provide a scientific basis on how the mode of action of ligands in heterogeneous catalysis can be rationalized, with the hope to promote this rather novel research area and to give some perspectives what may actually be feasible.
2 Changing Surface Properties of Supported Nanoparticles with Ligands
2.1 Theoretical Background and Representative Studies
As discussed above, the common approaches to alter the selectivity of a supported nanoparticle catalyst focus on modifying the surface properties of the particle. If we in a first approximation consider the organic tail of the ligand to be of minor importance, we can treat the influence of a ligand on the catalytic properties as a surface modification. The effect of a ligand on the surface can then be proposed to be twofold:
-
Saturating the surface atom to which it binds.
-
Change of the electronic structure of this surface atom.
Through binding a ligand, the free coordination site of a surface atom becomes blocked. This eliminates the ability of the surface atom to act as an adsorption site and therefore to activate and convert reactants. As a ligand may either withdraw or donate electron density, it has to be considered that it will alter the electronic structure of the metal to which it binds. An influence on the electronic properties may however not be restricted to the surface atom to which the ligand actually binds, but may further also alter atoms that are surrounding the ligand-blocked site. As a result the catalytic properties of such adjacent, ligand-free surface atoms may be changed, too.
By rationalizing the influence of ligands on a metal surface in this way, enables for discussing catalytic effects in a similar way as for bimetallic materials. Basically, two models have been established over the last decades in bimetallic catalysis that have been successfully applied in many cases:
-
Ensemble or geometric effect
-
electronic or ligand effect
An ensemble or geometric effect occurs when two metals of very different catalytic properties are mixed or more precisely, when mixing a metal that is a very active for a considered reaction with a metal that is very inactive for the same reaction. Starting from a surface that consists only of the active metal and replacing surface atoms step by step with atoms of the inactive metal leads to dilution of the highly active metal (see Fig. 3). As a result the number of adjacent active metal atoms decreases and thus the size of ensembles of contiguous active metal atoms. When comparing two reactions that can proceed in parallel but exhibit different ensembles size requirements, this means that for the reaction that needs larger ensembles the rate of reaction declines more strongly when increasing the amount of the inactive metal than for the reaction that requires smaller ensembles. As a result the overall selectivity for the reaction that proceeds over smaller ensembles is enhanced. Probably, the most famous example is the hydrogenolysis of C–C bonds and dehydrogenation of such bonds to form C=C over bimetallic Ni–Cu catalysts [49, 50]. While the rate of hydrogenolysis decreases significantly as the Cu content is increased, the rate of dehydrogenation remains constant over a wide compositional range. Ni is known to be catalytically active for both reactions whereas Cu in comparison to Ni can be considered as being almost catalytically inactive. As C–C hydrogenolysis requires large ensembles of adjacent catalytically active metal atoms, the rate of C–C scissoring decreases significantly when increasing the Cu contents. In contrast, dehydrogenation is considered to occur on small size ensembles. Therefore, the rate of dehydrogenation remains almost unaffected over a certain compositional range when diluting Ni with Cu.

Adding a second metal causes dilution of larger ensembles (ensemble or geometric effect). As a result smaller ensembles are formed at the expense of larger. Therefore, molecules that require large ensembles to adsorb cannot be activated and converted if the second metal is catalytically inactive for the considered reaction, as indicated by the red cross. The binding of ligands can lead to the same effect, because surface atoms which bind a ligand can be considered as being blocked and thus be catalytically inactive. A second metal can also donate electrons to the adjacent atoms or withdraw (electronic effect), as indicated by the brown arrow. Through binding a ligand to a surface atom the electronic structure of this atom is also altered. This may furthermore have an influence on the metal atoms adjacent to the ligand-blocked atom and therefore lead to a modification of their electronic and catalytic properties
Another more illustrative example to be worth mentioned is the hydrogenation of benzene and cyclohexene over supported bimetallic Pt–Au nanoparticles. Au in comparison to Pt can be considered as being catalytically inactive for the hydrogenation of unsaturated hydrocarbons. On pure Pt both, benzene and cyclohexene can be hydrogenated (see top part of Fig. 4). However, benzene requires larger ensembles for adsorption and activation than an isolated C=C bond [51]. As a result, the rate of hydrogenation for benzene over bimetallic Pt–Au nanoparticles with high Au contents diminishes completely whereas the same catalyst is still able to hydrogenate cyclohexene (see lower part of Fig. 4) [52].

Hydrogenation of benzene (left) and cyclohexene (right) over pure Pt (top) and bimetallic Pt–Au (bottom) surfaces. Pt atoms are blue and Au atoms yellow. Benzene requires larger ensembles to become adsorbed and activated than cyclohexene. As a result both can be hydrogenated on pure Pt (top). When diluting Pt with Au, which can be considered as being catalytically inactive for hydrogenation reactions, the large Pt ensembles are diluted at the expense of smaller. Therefore, benzene can no longer be effectively adsorbed and activated whereas cyclohexene hydrogenation still proceeds over such bimetallic catalysts
An “electronic or ligand effect” describes the scenario that the neighbors of an atom are substituted by atoms of a different metal. Therefore, the “ligand-sphere” is changed. This will alter the electronic structure of the atom and therefore its catalytic properties. In the field of heterogeneous bimetallic catalysis, the term ligand effect has been established for this electronic phenomenon [10]. In chemistry a ligand is however an organic molecule that is bound to a metal center. As discussed below ligands in heterogeneous catalysis do not merely lead to electronic but also to ensemble effects. Therefore, the description “ligand effect” may be somehow misleading and the term “electronic effect” should be used instead in order avoid any confusion.
The most impressive example for an electronic effect in bimetallic catalysis is the oxygen reduction by hydrogen over supported Pd–Au nanoparticles. A 50-fold increase of the turnover rate compared to the monometallic palladium sample was found when the Pd–Au ratio was tuned to about one to one. It was concluded that the presence of Au surrounding Pd alters the electronic properties of Pd, which ultimately decreases the oxygen binding energy.
If as mentioned above one considers a ligand to block and poison the surface atom to which it binds, its influence can be proposed to be comparable to the dilution of an active metal by an inactive one. Some examples for the use of ligands in heterogeneous catalysis reveal that this analogy seems indeed to be valid.
Furfurals are important model compounds for exploring how platform chemicals that have been derived from biomass feedstocks can be further converted. On bare Pd metal catalysts furfural preferentially undergoes decarbonylation and hydrogenation of the aromatic moiety (see reaction path a) in Fig. 5). In contrast, a desirable route is the chemoselective hydrogenation of the C=O bond and furthermore cleavage of the resulting C–O bond in order to convert oxygenates to potential fuel substitutes (see reaction path b) in Fig. 5). Using surface science techniques it has been shown that the undesired reaction path a) only proceeds if furfural is able to adsorb in a flat mode on the surface [53]. As a result this reaction path requires larger ensembles of adjacent surface atoms than the desired hydrogenation of the C=O bond. A dilution of larger ensembles should thus lead to an enhanced selectivity for the desired reaction pathway. IR spectroscopic studies using CO as a probe molecule and taking the ratio of linearly bound CO to CO in a bridge binding mode and hollow sites as a measure for the accessibility of small and large ensembles revealed that ligand binding leads to a shift of this ratio toward smaller ensembles [13, 54]. Catalytic tests performed with such catalysts demonstrated that the undesired reaction path can indeed be suppressed competently while the rate for the desired reaction is almost maintained [55]. These results hence suggest that the use of ligands in heterogeneous catalysis can indeed lead to ensemble effects similar as known from bimetallic catalysis, when diluting an active metal with an inactive one.

Furfural preferentially undergoes decarbonylation over Pd catalysts followed by hydrogenation of the aromatic moiety (see reaction path a). A desired reaction route is however the chemoselective hydrogenation of the C=O bond and cleavage of the resulting C–O bond (see reaction path b)
Examples for ligand induced electronic effects that alter the catalytic properties of supported nanoparticles have been demonstrated for the electrocatalytic oxygen reduction reaction (ORR) [56, 57]. Pt is known to be the best catalytic metal for ORR. The reaction however still exhibits a considerable overpotential that is determined by the binding of oxygenates [58]. The key to further enhance ORR activities is hence to lower the binding energies of such adsorbates. It has been demonstrated that ensembles are required for ORR [59]. Therefore, one would expect that anchoring ligands onto the surface of a Pt catalyst leads to significant activity losses, because the binding of ligands, as discussed above, causes dilution of larger ensembles [13, 54]. Instead a substantial activity enhancement was found when amine ligands such as oleylamine or aryl-ligands, were bound to supported Pt nanoparticles [56, 57]. This indicates the appearance of a strong electronic effect, presumably lowering the oxygenate binding energies that superimposes the activity losses induced by the dilution of ensembles, as overall a net activity enhancement is obtained by ligand functionalization. The underlying mechanistic aspects for these enhancement effects are however still under debate. In one work an electron donating effect from the ligand to the metal is proposed [56], whereas the authors of the other publication highlight the relevance of an electron withdrawing effect [57].
In fact it has to be considered that usually both, geometric and electronic effects occur in parallel. Only if one of the two effects significantly overwhelms the other it is possible to assign observed changes of the catalytic properties to either one of the two effects. An example that demonstrates the difficulty to differentiate between both effects is the hydrogenation of acetylene in ethylene rich streams, a reaction of significant industrial relevance [60, 61]. The challenge is to hydrogenate selectively acetylene to ethylene while inhibiting complete reduction toward ethane as well as the formation of oligomers that eventually poison the catalyst [62]. Industrially supported Pd catalysts are applied with CO used as a co-adsorbate. It has been shown that functionalization of supported Pd catalysts with ligands such as triphenyl phosphine or diphenyl sulfide leads to enhanced acetylene conversions, while decreasing the ability to hydrogenate ethylene. This results in an overall net gain in ethylene [63]. Furthermore, both ligands caused a suppression of the formation of undesired oligomers. Acetylene and ethylene hydrogenation rates were determined independently in order to gain further insights into the underlying mechanistic reasons for the enhanced selectivity. If the ligands merely act as blockers for surface atoms, one would actually expect both rates to decrease. The results however revealed that for both ligands only the ethylene hydrogenation rate was significantly reduced, whereas the acetylene hydrogenation rate remained almost completely unaffected [64]. This finding may hence be related to an electronic effect. However, also evidence for a geometric effect was demonstrated for both ligands using IR spectroscopy and CO as a probe molecule [64, 65]. Therefore, enhancement effects can hence not be clearly related to either one of the two effects. Instead it may be concluded that both act jointly leading to a synergistic effect.
2.2 Concluding Remarks, Limitations, and Unexplored Potentials of Ligands as Species to Alter Surface Properties
The above discussion demonstrates that the mechanism of action of ligands in heterogeneous catalysis can be rationalized to some extend by using models established in bimetallic catalysis. One should thus also discuss the potential of ligands with respect to bimetallic catalyst. A ligand-shell is lacking stability under harsher reaction conditions [66]. Their application must hence be concluded as limited to reactions that are performable under mild conditions such as hydrogenations. If selectivity losses occur by ligand-shell aging the catalyst can be regenerated by performing a subsequent ligand functionalization [67]. A common challenge for bimetallic catalysis is to achieve proper metal mixing and reproducible catalyst preparation [68]. With standard preparation techniques such as e.g. incipient wetness, the chemical composition often varies significantly from one particle to another, which may negatively affect the overall catalytic performance. In contrast, the preparation of ligand-functionalized particle catalysts is quite straight forward, as conventional supported particle catalysts have merely to be dispersed in a solution of the desired ligand [63]. Bimetallic materials undergo reconstruction when one reaction parameter changes. The actual surface composition and hence the catalytic properties are thus determined by the reaction conditions [26, 69]. This means that the catalysts will alter when the reaction conditions vary, as e.g. in a flow reactor, where the composition of the reactant stream changes across the bed length. Such reconstruction effects cannot occur for monometallic ligand-functionalized particles. It might hence be possible to maintain the ligand induced surface modification even under varying reaction conditions, as long as the ligand-shell is still stable under these conditions.
In the field of heterogeneous catalysis mainly amines and thiols have so far been explored as ligand anchoring groups. However, by taking a look at the homogeneous catalysis and self-assembling monolayer (SAM) literature, further inspiration can be found for anchoring groups. Of potential interest may be phosphorus based ligands, carbenes [70], nitriles [71], or silanes [72]. For colloidal Rh NPs phosphine and phosphite are effective stabilizers that allow for the application of such materials as “quasi-homogeneous” catalysts [33]. Recent studies showed that triphenylphosphite-functionalized Rh NPs enable for selective hydrogenation of styrene to ethylbenzene [73]. In contrast, Ru NPs functionalized with triphenylphosphine cause unselective hydrogenation of both the C=C bond and the aromatic moiety. Based on IR spectroscopic studies the enhanced selectivity was related to a preferential blocking of low-coordinated surface sites by the triphenylphosphite.
N-heterocyclic carbenes have been applied successfully as ligands for colloidal Ru and Pt NPs [74–76]. This class of ligands is of particular interest because N-heterocyclic carbenes bind to various transition metals [75]. They are less sensitive to oxidation than phosphines and many syntheses for various structures have been established over the last decades that enable for manipulating the steric and electronic properties [77]. Using different N-heterocyclic carbenes it was shown for Ru colloids, applied as “quasi-homogeneous” catalysts, that the activity for hydrogenation of aromatic compounds raises with increasing donor strength [74]. It can hence be concluded that by changing the electronic properties of carbenes their electronic effect on catalytic NPs can be tuned. This class of ligands may hence not merely be of potential interest for “quasi-homogeneous” NP catalysts, but also for supported NPs.
3 Ligand-Reactant Interactions on Supported Nanoparticles
3.1 Theoretical Background and Representative Studies
The primary function of the ligand in homogeneous metalorganic catalysis is to ensure stability of the catalytically active metal center as a complex that is dissolved in the reaction medium. Thereby, the interaction of the binding groups of the ligand with the metal center alters the electronic structure of the latter and thus its reactivity [78]. This effect can be considered as being similar to an electronic effect of ligands on the surface properties of nanoparticles, as discussed above.
There are however further potentials for ligands. As demonstrated in the field of homogeneous catalysis it is possible to achieve steric and electronic interactions between a ligand and a reactant. Electronic interactions can be established by non-covalent interactions such as hydrogen-bonding or π–π interactions [79, 80]. Such interactions are of great interest because they enable for manipulation of the reactant binding mode at the metal center and as a result to control selectivity. Furthermore, through ligand-reactant interactions alternative reaction mechanisms can be opened in which the ligand contributes to the reactant activation and conversion leading to rates that are enhanced in comparison to the purely metal-catalyzed reaction [81]. Such effects have thus been termed as “ligand-acceleration”. If ligand–reactant interactions are feasible on ligand-functionalized nanoparticles, this should enable for a completely different way of controlling selectivity. All established approaches such as using bimetallic materials, tuning particle size, or using promotors alter the geometric and electronic surface properties. These approaches can thus be summarized as “surface modifications”. The ability of achieving specific ligand–reactant interactions may however allow for the application of ligands as a selectivity controlling element perpendicular to the surface and therefore open a new and yet unexplored potential. A detailed look at the very recent literature on ligands in heterogeneous catalysis reveals that ligand–reactant interactions may indeed takes place on particle surfaces, as discussed in the following.
One of the greatest challenges for heterogeneous catalysis is stereoselectivity. As above mentioned, the use of specific alkaloids as chiral modifiers has been demonstrated as a suitable approach to tackle this issue. In order to obtain stereoselectivities it is however not necessary to apply a chiral molecules as a modifier. Some of the above discussed results on colloidal nanoparticles stabilized by chiral ligands and applied as “quasi-homogeneous” catalysts reveal that the chiral information can also be fixated to the surface in order to act as a ligand [34]. In order to address the challenge of stereoselectivity with ligands, it seems at first sight intuitive to use chiral ligands that are known from homogeneous catalysis to be tailored for achieving high stereoselectivities with specific reactants. This is unfortunately not possible, because the ligand and reaction medium properties in relation to each other play a crucial role for the use of ligands in heterogeneous catalysis. Stereoselective reactions are typically performed in organic media and ligands from homogeneous catalysis are designed to mediate the solubility of the catalyst in the reaction medium. Such ligands fulfill the same function when being applied to nanoparticles. The particles form a colloidal dispersion and the materials are hence are applicable as “quasi-homogeneous” catalysts [34]. They can however not be used as supported catalysts, because attractive ligand-reaction medium interactions cause particle desorption [82]. Therefore, the ligand and reaction medium have to be chosen so that the two do not mix. For reactions in nonpolar, organic media this allows for the use of small water soluble molecules such as amino acids and hence to use a huge pool of readily available, cheap, and harmless chiral ligands [46]. Alternatively one could use nonpolar ligands and a polar solvent.
The first asymmetric bias that has been achieved with such systems was obtained for the hydrogenation of 2-butanone (see Fig. 6) over cysteine- and cysteine-derivative-functionalized nanoparticle catalysts [46]. The stereoselectivities were very low (around 10 %). Within the past 2 years the use of different amino acids and according derivatives as ligands as well as testing other reactants has however already led to a raise of enantiomeric excess (ee) to around 34 % [83]. Recently, we have been available to achieve ees of about 63 % [84]. Thereby, proline has been identified as being privileged presumably due to its rigid structure, because steric interactions are known from homogeneous catalysis to be of significant importance for an asymmetric bias [85]. With regard to the evolution of stereoselective metalorganic catalysis [16, 85], the results so far obtained for ligand-functionalized nanoparticles encourage to continue this research in order to search for systems with stereoselectivities above 90 % ee.

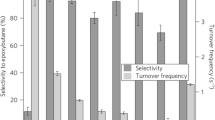
First but only low stereoselectivities have been obtained with supported Pt NPs functionalized with cysteine and cysteine derivatives for the hydrogenation of 2-butanone (a) [46]. The current benchmark when using hydrophilic ligands such as amino acids is 34 % achieved for the hydrogenation of ethylacetoacetate over proline-functionalized Pt NPs (b) [83]
Within homogeneous catalysis it is well accepted that the origin of any asymmetric bias lies in the steric and electronic interactions between chiral ligands and the adsorbed reactant, while the metal determines the catalytic activity [86]. When working with heterogeneous catalysts one has to question what the contribution of the surface within a steroselective reaction could be. For chiral modifiers the possibility of a “supramolecular chiral” environment formed by an ordered array of adsorbed modifiers has been proposed but eventually been excluded as the origin of stereoselectivity on the basis of surface science studies [38]. Instead stereoselectivity is related to the formation of a one to one modifier–reactant complex and the surface only to be required for providing the appropriate adsorption site and of course hydrogen activation. As discussed above, the adsorption mode of the modifier is critical for obtaining high stereoselectivities, but the asymmetric bias originates from the modifier–reactant interaction. Recent investigations on ligand-functionalized nanoparticles evidenced that the stereoselectivity does not depend on the particle size [83]. However, changes of the enantiomeric excess with regard to the ligand and its binding mode to the surface have been demonstrated [87]. These findings suggest that the surface structure is of minor relevance for the asymmetric bias in these systems, but the origin of the stereoselectivity is primarily related to the interaction between ligand and reactant, similar as in homogeneous catalysis.
Further evidence for the ability to control selectivity by ligand–reactant interactions is given by results obtained for the hydrogenation of cinnamaldehyde in ethanol over Al2O3 supported Pt nanoparticles functionalized with nonpolar thiols ligands [88]. The difficulty of this reaction (Fig. 7) is the chemoselective hydrogenation of the C=O bond in order to obtain the unsaturated alcohol (cinnamyl alcohol, green structure in Fig. 7).

Hydrogenation of cinnamaldehyde representative for α-β-unsaturated carbonyl compounds. Chemoselective hydrogenation of the C=O bond to the unsaturated alcohol (green structure) is the desired reaction. However, over typical hydrogenation catalysts the rate of hydrogenation for the C=C bond to give the red product is significantly higher. Eventually both groups are hydrogenated (grey product) for prolonged reaction periods
Over most hydrogenation catalysts such as Pt the rate of hydrogenation for the C=C bond is significantly higher than that for the C=O bond, which precludes obtaining high unsaturated alcohol yields [89]. A very interesting effect was observed when Al2O3-supported Pt particles were functionalized with phenylthiols of different CH2 spacers between the phenyl and the thiol group (see Fig. 8). A selectivity enhancement with a maximum of 90 % toward the unsaturated alcohol was obtained when the distance between the thiol and the phenyl group was increased to a spacer length of three CH2 groups [88]. Further increases of the distance between the two groups caused the selectivity then to decrease. This finding as well as the fact that alkylthiols did not lead to the same selectivity enhancements than phenylthiol ligands suggest that electronic and geometric effects on the surface properties, as described in 2.1, cannot hold as the sole explanation. Instead the enhancement effect is specific to the backbone of the ligand and was related to the formation of π–π interactions between the reactant and the ligand. This specific non-covalent interaction leads to an ideal orientation of cinnamaldehyde for hydrogenation of the C=O bond as illustrated in Fig. 8. These experimental findings evidence the potential of ligand–reactant interactions to steer selectivity in heterogeneous catalysis in a similar manner as known from homogeneous catalysis.

Phenyl thiol ligands of different spacing between the two functional groups have been tested as ligands for the hydrogenation of cinnamaldehyde over supported Pt nanoparticle catalysts [88]. A pronounced selectivity was obtained when the length of ligand was almost the same as the length of the reactant. This suggests a secondary electronic interaction between ligand and reactant via π–π-stacking as indicated on the right
One drawback of thiol ligands is their strong poisoning effect. Therefore, selectivity enhancements with thiol ligands are often accompanied by a loss of activity [46, 88]. Different observations have been made for the use of amine ligands. For instance, the use of l-proline (see Fig. 6b) as a simple amine ligand can lead to a simultaneous enhancements in activity and selectivity [82]. From homogeneous catalysis it is known that primary and secondary amines enable for an alternative reaction pathway in which the amine bound hydrogen substituents act as protons to activate the oxygen (N–H effect, see Fig. 9) [81]. The actual hydrogenation proceeds via transfer of the amine proton to the oxygen and hydride transfer from the metal to the carbon atom of the C=O group. This reaction pathway exhibits a higher rate of reaction than the purely metal catalyzed reaction, so that the ligand induced reaction causes an activity increase. Ligand induced activity enhancements are a well-known phenomenon in homogeneous catalysis and are related to as ligand-acceleration [4]. Also the above discussed chiral modifiers (see Fig. 1) exhibit a pronounced acceleration effect [42].

Hydrogenation of carbonyl compound via N–H assisted reaction pathway: hydrogen substituents of amines become acidic upon binding of the amine group to a catalytic late d-transition metals [81]. The oxygen of a C=O group can interact with such a proton and thereby get activated (step 1). Hydrogenation then occurs by transfer of the proton to the oxygen atom and hydride transfer from the metal to the carbon atom (step 2). Molecular hydrogen is subsequently coordinated (step 3) and heterogeneously dissociated (step 4), closing the catalytic cycle. Reproduced form reference 78 with permission from Royal Society of Chemistry
A simple test for the presence of the above described N–H effect is the use of tertiary amine [86]. As a tertiary amine does not exhibit any hydrogen substituents such ligands cannot generate an N–H effect and therefore must lead to an activity decrease, because they can merely act as a surface blocker. In the case of proline and N-methyl-proline the test for an N-H effect has been found to be positive for two reactions [82, 83]. This suggests that the amine-bound hydrogen plays an essential role for obtaining high activities. It should however be considered that the reaction cannot proceed over a single surface atom as in homogeneous catalysis and illustrated in Fig. 9. Instead the active site seems to consist of a site pair of a proline binding surface atom and an adjacent ligand-free one [83]. Besides that further studies will be necessary in order to clarify ligand-acceleration effects on nanoparticles in more detail, these results demonstrate that the catalytic activity of nanoparticles is not necessarily reduced by binding ligands. Instead it is possible to enhance activities by binding appropriate ligands to the surface which can be related to the presence of ligand-acceleration effects. Ligands may hence not just merely act as a spectator, but they can assist within or even change the reaction pathway as known from homogeneous catalysis.
The above given examples propose the relevance of specific ligand–reactant interactions as known from homogeneous catalysis. Another aspect of ligand–reactant interactions can be envisioned when considering the ligand shell as a 3-dimensional matrix. In this case the ligand shell could act as a porous filter through which specific molecules may diffuse to the surface to become activated while others are kept away. E.g. it has been shown recently that the form and order of the ligand shell depends on the ligand coverage [90]. At low coverages ligands with long alkyl chains cover the particle by lying flat on the surface to form a 2-dimension like coating. In contrast, at high coverages due to the limited space on the surface the ligands straighten up to form a 3-dimensional shell. This structural change was shown to alter the catalytic properties of supported Pd NPs for the semi-hydrogenation of alkynes to alkenes and the reactant diffusion through the ligand sphere was discussed as one essential aspect for this behavior. However, not only the coverage but also the ligand structure can alter the form of a ligand shell and thereby the catalytic properties. For instance the order of shells formed of alkyl ligands increases with increasing tail length and the activities of such systems for the hydrogenation of 1-epoxy-3-butene to 1-epoxy-3-butane was found to correlate with the order of the ligand shell [91, 92]. This evidences that the order of a ligand shells is one important aspect that influences the accessibility of the catalytic surface.
3.2 Conclusion and Outlook for the Application of Ligand–Reactant Interactions on Supported Particle Catalysts
As discussed in 3.1 and also demonstrated by further examples [93], it is basically possible to achieve specific, non-covalent ligand–reactant interactions, similar as in the field of homogenous catalysis. This opens a completely new perspective for heterogeneous catalysis with yet unknown potentials that have to be explored within the next years. Most of the early investigations on the use of ligands in heterogeneous catalysis show strong inspiration from the field of SAMs and focused mainly on the use of alkylthiols and alkylamines [1, 91]. Such ligands have been demonstrated to be suitable and effective for altering the surface properties. Furthermore, they can alter the accessibility of the surface by acting as a 3-dimensional matrix through which the reactant has to diffuse. When trying to establish and apply specific ligand–reactant interactions such ligands have however to be considered as “inert”. Instead ligands should rather exhibit rigid skeletons and functional groups (e.g. phenyl, OH). This enables for achieving steric and electronic ligand–reactant interactions that pave the way to manipulate the adsorption, activation, and even the reaction pathway of a reactant on a molecular level. Only a very few but striking examples have been reported yet that demonstrate this potential. This evidences that there is plenty of space for new discoveries when imagining a surface-bound ligand to be utilizable in a similar way as ligands in homogeneous catalysis.
Finally, the idea of using a ligand not as a passive adsorbate but as a species with chemical properties that may be useful to tune the catalytic properties is not restricted to thermally driven reactions in heterogeneous catalysis. A recent example from the field of electrocatalysis has shown that it is possible to bind electrochemically active ligands to Pt nanoparticles and to maintain their redox activity [94]. For future studies it would be interesting to continue this approach in order to explore the possibility of achieving cooperative electrocatalytic effects between ligand and nanoparticle.
4 Concluding Remarks
The possibility of ligands to tune the catalytic properties of heterogeneous catalysts has been overlooked for quite some time and their ability to act as poisoning species by blocking the catalytic surface of supported nanoparticle was usually given as the main reason [1]. It has however in recent years been demonstrated that ligands can be used to block undesired reaction pathways while maintaining the formation rate toward the desired product. This enables for tuning the overall selectivity. Furthermore, by achieving ligand-acceleration effects on nanoparticles it is not merely possible to enhance selectivity but also activity simultaneously. Within the light of these and further recently published examples the “traditional way of thinking” regarding ligands in heterogeneous catalysts has to be revalued and intensive research will be necessary to reveal their full potential to tune the catalytic properties of supported nanoparticle catalysts. With regard to this task the present article attempts to give some guidance for future investigations in order to promote the progress in this emerging research field.
References
Sonstrom P, Baumer M (2011) Supported colloidal nanoparticles in heterogeneous gas phase catalysis: on the way to tailored catalysts. Phys Chem Chem Phys 13:19270–19284
Tolman CA (1977) Steric effects of phosphorus ligands in organometallic chemistry and homogeneous catalysis. Chem Rev 77:313–348
Komarov IV, Borner A (2001) Highly enantioselective or not?—Chiral monodentate monophosphorus ligands in the asymmetric hydrogenation. Angew Chem Int Ed 40:1197–1200
Berrisford DJ, Bolm C, Sharpless KB (1995) Ligand-accelerated catalysis. Angew Chem Int Ed 34:1059–1070
DOE (Departement of Energy) (2007) Basic research needs: catalysis for energy. United States Department of Energy, Washington DC
Roadmap for Catalysis Research in Germany (2010) Roadmap for Catalysis Research in Germany German Catalysis Society (GeCatS)
Somorjai GA, McCrea K (2001) Roadmap for catalysis science in the 21st century: a personal view of building the future on past and present accomplishments. Appl Catal A 222:3–18
Bezemer GL, Bitter JH, Kuipers H, Oosterbeek H, Holewijn JE, Xu XD, Kapteijn F, van Dillen AJ, de Jong KP (2006) Cobalt particle size effects in the Fischer–Tropsch reaction studied with carbon nanofiber supported catalysts. J Am Chem Soc 128:3956–3964
Galvagno S, Capannelli G, Neri G, Donato A, Pietropaolo R (1991) Hydrogenation of cinnamaldehyde over Ru/C catalysts: effect of Ru particle size. J Mol Catal 64:237–246
Sinfelt JH (1983) Bimetallic catalysts: discoveries, concepts, and applications. Wiley, New York
Edwards JK, Hutchings GJ (2008) Palladium and gold–palladium catalysts for the direct synthesis of hydrogen peroxide. Angew Chem Int Ed 47:9192–9198
Englisch M, Jentys A, Lercher JA (1997) Structure sensitivity of the hydrogenation of crotonaldehyde over Pt/SiO2 and Pt/TiO2. J Catal 166:25–35
Pang SH, Schoenbaum CA, Schwartz DK, Medlin JW (2013) Directing reaction pathways by catalyst active-site selection using self- assembled monolayers. Nat Commun 4:2448
Witte PT, Berben PH, Boland S, Boymans EH, Vogt D, Geus JW, Donkervoort JG (2012) BASF nanoSelect (TM) technology: innovative supported pd- and pt-based catalysts for selective hydrogenation reactions. Top Catal 55:505–511
Noyori R (2002) Asymmetric catalysis: science and opportunities (nobel lecture). Angew Chem Int Ed 41:2008–2022
Knowles WS (2003) Asymmetric hydrogenations (nobel lecture 2001). Adv Synth Catal 345:3–13
Bonnemann H, Richards RM (2001) Nanoscopic metal particles—synthetic methods and potential applications. Eur J Inorg Chem 10:2455–2480
Hirai H, Nakao Y, Toshima N (1978) Preparation of colloidal rhodium in poly(vinyl alcohol) by reduction with methanol. J Macromol Sci Chem A12:1117–1141
Hirai H, Nakao Y, Toshima N (1979) Preparation of colloidal transition-metals in polymers by reduction with alcohols or ethers. J Macromol Sci Chem A13:727–750
Comotti M, Li WC, Spliethoff B, Schuth F (2006) Support effect in high activity gold catalysts for CO oxidation. J Am Chem Soc 128:917–924
Reetz MT, Winter M, Breinbauer R, Thurn-Albrecht T, Vogel W (2001) Size-selective electrochemical preparation of surfactant-stabilized Pd-, Ni- and Pt/Pd colloids. Chem Eur J 7:1084–1094
Toshima N, Yonezawa T (1998) Bimetallic nanoparticles—novel materials for chemical and physical applications. New J Chem 22:1179–1201
Tamura M, Fujihara H (2003) Chiral bisphosphine BINAP-stabilized gold and palladium nanoparticles with small size and their palladium nanoparticle-catalyzed asymmetric reaction. J Am Chem Soc 125:15742–15743
Weare WW, Reed SM, Warner MG, Hutchison JE (2000) Improved synthesis of small (d(CORE) approximate to 1.5 nm) phosphine-stabilized gold nanoparticles. J Am Chem Soc 122:12890–12891
Huang WX, Hua Q, Cao T (2014) Influence and removal of capping ligands on catalytic colloidal nanoparticles. Catal Lett 144:1355–1369
Kunz S, Iglesia E (2014) Mechanistic evidence for sequential displacement-reduction routes in the synthesis of Pd–Au clusters with uniform size and clean surfaces. J Phys Chem C 118:7468–7479
Aliaga C, Park JY, Yamada Y, Lee HS, Tsung CK, Yang PD, Somorjai GA (2009) Sum frequency generation and catalytic reaction studies of the removal of organic capping agents from Pt nanoparticles by UV-ozone treatment. J Phys Chem C 113:6150–6155
Rioux RM, Hsu BB, Grass ME, Song H, Somorjai GA (2008) Influence of particle size on reaction selectivity in cyclohexene hydrogenation and dehydrogenation over silica-supported monodisperse Pt particles. Catal Lett 126:10–19
Bratlie KM, Lee H, Komvopoulos K, Yang P, Somorjai GA (2007) Platinum nanoparticle shape effects on benzene hydrogenation selectivity. Nano Lett 7:3097–3101
Altmann L, Wang X, Borchert H, Kolny-Olesiak J, Zielasek V, Parisi J, Kunz S, Baumer M (2015) Influence of Sn content on the hydrogenation of crotonaldehyde catalysed by colloidally prepared PtSn nanoparticles. Phys Chem Chem Phys 17:28186–28192
Narayanan R, El-Sayed MA (2005) Catalysis with transition metal nanoparticles in colloidal solution: nanoparticle shape dependence and stability. J Phys Chem B 109:12663–12676
González-Gálvez D, Nolis P, Philippot K, Chaudret B, van Leeuwen PWNM (2012) Phosphine-stabilized ruthenium nanoparticles: the effect of the nature of the ligand in catalysis. ACS Catal 2:317–321
Castelbou JL, Gual A, Mercade E, Claver C, Godard C (2013) Ligand effect in the Rh–NP catalysed partial hydrogenation of substituted arenes. Catal Sci Technol 3:2828–2833
Sawai K, Tatumi R, Nakahodo T, Fujihara H (2008) Asymmetric Suzuki–Miyaura coupling reactions catalyzed by chiral palladium nanoparticles at room temperature. Angew Chem Int Ed 47:6917–6919
Holland MC, Meemken F, Baiker A, Gilmour R (2015) Chiral imidazolidinone and proline-derived surface modifiers for the Pt-catalysed asymmetric hydrogenation of activated ketones. J Mol Catal A 396:335–345
Ma Z, Zaera F (2005) Role of the solvent in the adsorption-desorption equilibrium of cinchona alkaloids between solution and a platinum surface: correlations among solvent polarity, cinchona solubility, and catalytic performance. J Phys Chem B 109:406–414
Blaser HU, Studer M (2007) Cinchona-modified platinum catalysts: from ligand acceleration to technical processes. Acc Chem Res 40:1348–1356
Mallat T, Orglmeister E, Baiker A (2007) Asymmetric catalysis at chiral metal surfaces. Chem Rev 107:4863–4890
Kubota J, Zaera F (2001) Adsorption geometry of modifiers as key in imparting chirality to platinum catalysts. J Am Chem Soc 123:11115–11116
Hess R, Vargas A, Mallat T, Bürgi T, Baiker A (2004) Inversion of enantioselectivity in the platinum-catalyzed hydrogenation of substituted acetophenones. J Catal 222:117–128
Ferri D, Burgi T (2001) An in situ attenuated total reflection infrared study of a chiral catalytic solid-liquid interface: cinchonidine adsorption on Pt. J Am Chem Soc 123:12074–12084
Blaser HU, Jalett HP, Monti DM, Baiker A, Wehrli JT (1991) Enantioselective hydrogenation of ethyl pyruvate: effect of catalyst and modifier structure. In: Grasselli RK, Sleight AW (eds) Studies in surface science and catalysis. Elsevier, Amsterdam, pp 147–155
Blaser HU, Jalett HP, Lottenbach W, Studer M (2000) Heterogeneous enantioselective hydrogenation of ethyl pyruvate catalyzed by cinchona-modified Pt catalysts: effect of modifier structure. J Am Chem Soc 122:12675–12682
Mori A, Miyakawa Y, Ohashi E, Haga T, Maegawa T, Sajiki H (2006) Pd/C-catalyzed chemoselective hydrogenation in the presence of diphenylsulfide. Org Lett 8:3279–3281
Weng Z, Zaera F (2014) Increase in activity and selectivity in catalysis via surface modification with self-assembled monolayers. J Phys Chem C 118:3672–3679
Kunz S, Schreiber P, Ludwig M, Maturi MM, Ackermann O, Tschurl M, Heiz U (2013) Rational design, characterization and catalytic application of metal clusters functionalized with hydrophilic, chiral ligands: a proof of principle study. Phys Chem Chem Phys 15:19253–19261
Murzin DY, Maki-Arvela P, Toukoniitty E, Salmi T (2005) Asymmetric heterogeneous catalysis: science and engineering. Catal Rev 47:175–256
Bartok M (2006) Heterogeneous catalytic enantioselective hydrogenation of activated ketones. Curr Org Chem 10:1533–1567
Sinfelt JH, Carter JL, Yates DJC (1972) Catalytic hydrogenolysis and dehydrogenation over copper-nickel alloys. J Catal 24:283–296
Ponec V, Sachtler WMH (1972) The reactions between cyclopentane and deuterium on nickel and nickel-copper alloys. J Catal 24:250–261
Campbell JM, Seimanides S, Campbell CT (1989) Probing ensemble effects in surface reactions. 2. Benzene adsorption on clean and bismuth-covered platinum(111). J Phys Chem 93:815–826
Puddu S, Ponec V (1976) Effect of the ensemble size in the hydrogenation of benzene on Pt and Pt–Au catalysts. Recl Trav Chim Pays-Bas 95:255–257
Pang SH, Medlin JW (2011) Adsorption and reaction of furfural and furfuryl alcohol on Pd(111): unique reaction pathways for multifunctional reagents. ACS Catal 1:1272–1283
Altmann L, Kunz S, Bäumer M (2014) Influence of organic amino and thiol ligands on the geometric and electronic surface properties of colloidally prepared platinum nanoparticles. J Phys Chem C 118:8925–8932
Pang SH, Schoenbaum CA, Schwartz DK, Medlin JW (2014) Effects of thiol modifiers on the kinetics of furfural hydrogenation over Pd catalysts. ACS Catal 4:3123–3131
Chung Y-H, Chung DY, Jung N, Sung Y-E (2013) Tailoring the electronic structure of nanoelectrocatalysts induced by a surface-capping organic molecule for the oxygen reduction reaction. J Phys Chem Lett 4:1304–1309
Zhou Z-Y, Kang X, Song Y, Chen S (2012) Ligand-mediated electrocatalytic activity of Pt nanoparticles for oxygen reduction reactions. J Phys Chem C 116:10592–10598
Nørskov JK, Rossmeisl J, Logadottir A, Lindqvist L, Kitchin JR, Bligaard T, Jónsson H (2004) Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J Phys Chem B 108:17886–17892
Siahrostami S, Verdaguer-Casadevall A, Karamad M, Deiana D, Malacrida P, Wickman B, Escudero-Escribano M, Paoli EA, Frydendal R, Hansen TW, Chorkendorff I, Stephens IEL, Rossmeisl J (2013) Enabling direct H2O2 production through rational electrocatalyst design. Nat Mater 12:1137–1143
Borodzinki A (2006) Selective hydrogenation of ethyne in ethene-rich streams on palladium catalysts. Part 1. Effect of changes to the catalyst during reaction. Catal Rev 48:91–144
Borodzinski A, Bond GC (2008) Selective hydrogenation of ethyne in ethene-rich streams on palladium catalysts, part 2: steady-state kinetics and effects of palladium particle size, carbon monoxide, and promoters. Catal Rev 50:379–469
Bos ANR, Westerterp KR (1993) Mechanism and kinetics of the selective hydrogenation of ethyne and ethene. Chem Eng Process 32:1–7
McKenna FM, Mantarosie L, Wells RPK, Hardacre C, Anderson JA (2012) Selective hydrogenation of acetylene in ethylene rich feed streams at high pressure over ligand modified Pd/TiO2. Catal Sci Technol 2:632–638
McKenna F-M, Wells RPK, Anderson JA (2011) Enhanced selectivity in acetylene hydrogenation by ligand modified Pd/TiO2 catalysts. Chem Commun 47:2351–2353
McCue AJ, McKenna F-M, Anderson JA (2015) Triphenylphosphine: a ligand for heterogeneous catalysis too? Selectivity enhancement in acetylene hydrogenation over modified Pd/TiO2 catalyst. Catal Sci Technol 5:2449–2459
Wang X, Sonstrom P, Arndt D, Stover J, Zielasek V, Borchert H, Thiel K, Al-Shamery K, Baumer M (2011) Heterogeneous catalysis with supported platinum colloids: a systematic study of the interplay between support and functional ligands. J Catal 278:143–152
Kahsar KR, Schwartz DK, Medlin JW (2015) Stability of self-assembled monolayer coated Pt/Al2O3 catalysts for liquid phase hydrogenation. J Mol Catal A 396:188–195
Sachdev A, Schwank J (1989) Microstructure and reactivitiy of supported bimetallic platinum gold catalysts. J Catal 120:353–369
Tao F, Grass ME, Zhang YW, Butcher DR, Renzas JR, Liu Z, Chung JY, Mun BS, Salmeron M, Somorjai GA (2008) Reaction-driven restructuring of Rh–Pd and Pt–Pd core–shell nanoparticles. Science 322:932–934
Herrmann WA, Goossen LJ, Kocher C, Artus GRJ (1996) Chiral heterocyclic carbenes in asymmetric homogeneous catalysis. Angew Chem Int Ed Engl 35:2805–2807
Rach SF, Kuehn FE (2009) Nitrile ligated transition metal complexes with weakly coordinating counteranions and their catalytic applications. Chem Rev 109:2061–2080
Long Y-T, Herrwerth S, Eck W, Grunze M (2002) Synthesis and characterization of self-assembled monolayers based on redox-active silane compounds on platinum surfaces. Phys Chem Chem Phys 4:522–526
Castelbou JL, Blondeau P, Claver C, Godard C (2015) Surface characterisation of phosphine and phosphite stabilised Rh nanoparticles: a model study. RSC Adv 5:97036–97043
Gonzalez-Galvez D, Lara P, Rivada-Wheelaghan O, Conejero S, Chaudret B, Philippot K, van Leeuwen PWNM (2013) NHC-stabilized ruthenium nanoparticles as new catalysts for the hydrogenation of aromatics. Catal Sci Technol 3:99–105
Lara P, Rivada-Wheelaghan O, Conejero S, Poteau R, Philippot K, Chaudret B (2011) Ruthenium nanoparticles stabilized by N-heterocyclic carbenes: ligand location and influence on reactivity. Angew Chem Int Ed 50:12080–12084
Lara P, Suarez A, Colliere V, Philippot K, Chaudret B (2014) Platinum N-heterocyclic carbene nanoparticles as new and effective catalysts for the selective hydrogenation of nitroaromatics. ChemCatChem 6:87–90
Díez-González S, Marion N, Nolan SP (2009) N-Heterocyclic carbenes in late transition metal catalysis. Chem Rev 109:3612–3676
Clarke ML, Frew JJR (2009) Ligand electronic effects in homogeneous catalysis using transition metal complexes of phosphine ligands. The Royal Society of Chemistry, Cambridge
Borner A (2001) The effect of internal hydroxy groups in chiral diphosphane rhodium(I) catalysts on the asymmetric hydrogenation of functionalized olefins. Eur J Inorg Chem 2:327–337
Sawamura M, Ito Y (1992) Catalytic asymmetric synthesis by means of secondary interaction between chiral ligands and substrates. Chem Rev 92:857–871
Clapham SE, Hadzovic A, Morris RH (2004) Mechanisms of the H2-hydrogenation and transfer hydrogenation of polar bonds catalyzed by ruthenium hydride complexes. Coord Chem Rev 248:2201–2237
Schrader I, Warneke J, Backenköhler J, Kunz S (2015) Functionalization of platinum nanoparticles with l-proline: simultaneous enhancements of catalytic activity and selectivity. J Am Chem Soc 137:905–912
Schrader I, Neumann S, Himstedt R, Zana A, Warneke J, Kunz S (2015) The effect of particle size and ligand configuration on the asymmetric catalytic properties of proline-functionalized Pt-nanoparticles. Chem Commun 51:16221–16224. doi:10.1039/C1035CC06990D
Schrader I, Neumann S, Schmidt F, Feige F, Kunz S (2016) Publication in preparation
Noyori R (2003) Asymmetric catalysis: science and opportunities (nobel lecture 2001). Adv Synth Catal 345:15–32
Noyori R, Ohkuma T (2001) Asymmetric catalysis by architectural and functional molecular engineering: practical chemo- and stereoselective hydrogenation of ketones. Angew Chem Int Ed 40:40–73
Kunz S, Maturi MM, Schrader I, Backenköhler J, Tschurl M, Heiz U (2014) Same ligand—different binding mode: a way to control the binding of N-acetyl-cysteine (NAC) to Pt clusters. J Colloid Interface Sci 426:264–269
Kahsar KR, Schwartz DK, Medlin JW (2014) Control of metal catalyst selectivity through specific noncovalent molecular interactions. J Am Chem Soc 136:520–526
Gallezot P, Richard D (1998) Selective hydrogenation of alpha, beta-unsaturated aldehydes. Catal Rev 40:81–126
Albani D, Vile G, Mitchell S, Witte PT, Almora-Barrios N, Verel R, Lopez N, Perez-Ramirez J (2016) Ligand ordering determines the catalytic response of hybrid palladium nanoparticles in hydrogenation. Catal Sci Technol 6:1621–1631
Marshall ST, O’Brien M, Oetter B, Corpuz A, Richards RM, Schwartz DK, Medlin JW (2010) Controlled selectivity for palladium catalysts using self-assembled monolayers. Nat Mater 9:853–858
Marshall ST, Schwartz DK, Medlin JW (2011) Adsorption of oxygenates on alkanethiol-functionalized Pd(111) surfaces: mechanistic Insights into the role of self-assembled monolayers on catalysis. Langmuir 27:6731–6737
Makosch M, Lin W-I, Bumbálek V, Sá J, Medlin JW, Hungerbühler K, van Bokhoven JA (2012) Organic thiol modified Pt/TiO2 catalysts to control chemoselective hydrogenation of substituted nitroarenes. ACS Catal 2:2079–2081
Morsbach E, Nesselberger M, Warneke J, Harz P, Arenz M, Baumer M, Kunz S (2015) 1-Naphthylamine functionalized Pt nanoparticles: electrochemical activity and redox chemistry occurring on one surface. New J Chem 39:2557–2564
Acknowledgments
The author gratefully acknowledges financial support through a Liebig Habilitations Stipendium of the “Fonds der Chemischen Industrie”. Many thanks to Dr. Volkmar Zielasek and Imke Schrader for careful proofreading and thoughtful suggestions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kunz, S. Supported, Ligand-Functionalized Nanoparticles: An Attempt to Rationalize the Application and Potential of Ligands in Heterogeneous Catalysis. Top Catal 59, 1671–1685 (2016). https://doi.org/10.1007/s11244-016-0687-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-016-0687-7