
Abstract
Three new coordination polymers, namely, {[Cu2(bcpmba)(μ4-OH)]·2H2O}n (1), [Mn(Hbcpmba)]n (2), and [Co2(bcpmba)(μ3-OH)·H2O]n (3) (H3bcpmba = 3,5-bi(4-carboxy-phenylene-methylene-oxy)-benzoic acid) have been prepared under solvothermal conditions. The complexes were characterized by physico-chemical and spectroscopic methods. All of the compounds 1–3 contain one-dimensional (1D) chains extended via the bcpmba3− bridge to generate 2D porous layers which are further connected by bcpmba3− ligands to form 3D porous coordination polymers. The result shows configurations of the ligand have an important influence on the structure. Magnetic susceptibility measurements indicate that compounds 1–3 exhibit antiferromagnetic coupling between adjacent metal ions, with the corresponding J value of − 2.76 cm−1 for compound 2.
Graphic abstract
Three porous coordination polymers, namely, {[Cu2(bcpmba)(μ4-OH)]·2H2O}n (1), [Mn(Hbcpmba)]n (2) and [Co2(bcpmba)(μ3-OH)·H2O]n (3) have been synthesized by employing a semi-rigid aromatic multicarboxylate acid (3,5-bi(4-carboxy-phenylene-methylene-oxy)-benzoic acid, H3bcpmba) under solvothermal conditions. Porous coordination polymers 1–3 consisted of 1D chain extended via the bridge of bcpmba3– to generate 2D porous layers and further connected by bcpmba3– to provide a 3D porous frameworks. The results reveal that different coordination modes of the ligand play an important role in the self-assembly processes to form metal-organic frameworks with different structures. Moreover, compounds 1–3 exhibited antiferromagnetic properties.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The design and construction of the porous coordination polymers (PCPs) are extremely attractive, due to their potential applications in gas storage, separation, heterogeneous catalysis, and magnetism [1, 2]. Organic linkers and metal centers are of vital importance in the design and synthesis of porous coordination polymers with the expected structure and properties [3]. Among a large variety of organic building blocks utilized for constructing new PCPs and related compounds, aromatic multicarboxylic acids represent a particularly promising class of ligands owing to their high coordination versatility, different levels of deprotonation, thermal stability, and suitability for hydrothermal synthesis [4,5,6,7,8]. The semirigid aromatic multicarboxylate ligands are still good candidates for building PCPs on account of their rich coordination modes and various conformations [9, 10]. In the process of building PCPs, the semirigid aromatic multicarboxylate ligands can relatively easily regulate its configurations to enhance the structural diversity, in contrast to the rigid ligands with difficult conformational changes [11, 12].
Based on the above-mentioned considerations, a semirigid tripodal carboxylate ligand, H3bcpmba (Scheme 1), was selected to synthesize coordination polymers with various architectures. Therefore, H3bcpmba was selected as the building block based on the following considerations: First, three benzene rings in H3bcpmba are rigid, therefore they can accommodate space in a coordination polymer, thus facilitating the formation of porous coordination polymers [13]. Second, the additional –O– spacer in H3bcpmba rotates and bends to adopt various configurations, thus exhibiting flexible nature [14]. Third, three carboxylic groups provide a magnetic superexchange between the metal centers [14]; therefore, materials with excellent properties may be obtained using H3bcpmba.

Structural formulae of ligand used in this work
In this study, three new coordination polymers, namely, {[Cu2(bcpmba)(μ4-OH)]·2H2O}n (1), [Mn(Hbcpmba)]n (2) and [Co2(bcpmba)(μ3-OH)·H2O]n (3) were synthesized under hydrothermal conditions. Three new compounds have been characterized by elemental analysis, IR spectra, TG, and X-ray crystallography. The crystal structures, magnetic properties, and thermal properties are studied in detail.
Experimental section
General remarks
All reagents were from commercial sources (analytical reagent grade) and were used as received. H3bcpmba ligand was purchased from Jinan Henghua Sci. & Tec. Co. Ltd. Infrared spectra were measured on a Nicolet Avatar 360 FTIR instrument from 4000 to 400 cm−1 using KBr pellets. Elemental analyses of C, H, and N were carried out on a Perkin-Elemer 2400 CHNS Elemental Analyzer. Powder X-ray diffraction was performed on a Bruker D8 ADVANCE X-ray powder diffractometer with Cu Kα radiation (λ = 1.5406 Å). Thermogravimetric analyses were performed on a Netzsch TG209F3 thermogravimetric analyzer from 30 to 900 °C at 5 °C min−1 under N2. The variable-temperature magnetic susceptibility measurements for polycrystalline complexes 1–3 were collected on a Quantum Design SQUID MPMS XL-7 instrument over the temperature range of 2–300 K under an applied field of 1000 Oe, and the diamagnetic corrections were evaluated by using Pascal’s constants.
Preparation of {[Cu2(bcpmba)(μ4-OH)]·2H2O}n (1)
A mixture of CuCl2·4H2O (17 mg, 0.1 mmol) and H3bcpmba (21 mg, 0.05 mmol) was added to a mixed solvent system containing acetonitrile (4 mL) and water (2 mL). The contents were first sealed in a 25 mL Teflon-lined stainless vessel and heated at 160 °C for 72 h and then gradually cooled to room temperature at a rate of 5 °C h−1. Blue colored rod-like crystals of 1 were collected by filtration and washed with methanol and ethanol. Yield 16 mg (74%) based on H3bcpmba. Calcd. (Found) for C11.5H8CuO4.5: C, 49.25 (49.21); H, 2.84 (2.80); N, 0.00 (0.00) %. IR (KBr pellet, cm−1): 3421 (m), 2981 (m), 1581 (m), 1411 (s), 1151 (m), 966 (w), 769 (m), 675 (w), 457 (w).
Preparation of [Mn(Hbcpmba)]n (2)
The procedure was similar to the preparation of 1, except that CuCl2·4H2O was replaced with MnCl2·4H2O. Yield 10 mg (49%) based on H3bcpmba. Calcd. (Found) for C23H16MnO8: C, 58.19 (58.12); H, 3.37 (3.30); N, 0.00 (0.00) %. IR (KBr pellet, cm−1): 3440 (w), 2879 (w), 2499 (w), 1599 (s), 1369 (s), 1159 (s), 924 (w), 773 (m), 707 (w), 528 (w).
Preparation of [Co2(bcpmba)(μ3-OH)·H2O]n (3)
The procedure was similar to the preparation of 1, except that CuCl2·4H2O was replaced with CoCl2·6H2O. Yield 28 mg (47%) based on H3bcpmba. Calcd. (Found) for C23H18Co2O11: C, 46.92 (46.94); H, 3.06 (3.04); N, 0.00 (0.00) %. IR (KBr pellet, cm−1): 3424 (m), 1550 (s), 1405 (s), 1155 (m), 1054 (w), 769 (m).
Crystal structure determination
Crystallographic data for the compounds were collected on a Bruker Apex Smart CCD diffractometer with graphite-monochromated Mo–Ka radiation (λ = 0.71073 Å) using the ω-scan technique at room temperature. SAINT software was used for data integration and reduction [15]. Absorption correction was performed with SADABS [16]. All the structures were solved by the direct method employing SHELXS-97 and SHELXL-97 was used to refine on F2 with full-matrix least squares technique [17, 18]. The non-hydrogen atoms were refined with the anisotropic displacement parameters. The hydrogen atoms were set in calculated positions and refined as riding atoms with a common isotropic thermal parameter. The contribution of the electron density by the remaining disorder solvent molecule in the channels of was removed by the SQUEEZE routine in PLATON [19].
Details of the crystal parameters, data collections and refinement for 1–3 are summarized in Table 1. Selected bond lengths and angles with their estimated standard deviations are given in Table 2.
Results and discussion
Structural description of the complexes
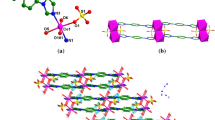
{[Cu2(bcpmba)(μ4-OH)]·2H2O}n (1). Compound 1 crystallizes in the monoclinic space group C2/c and generates a 3D porous coordination polymer. The asymmetric unit consists of one copper atom, one half of a completely deprotonated bcpmba3− ligand, one half of the μ4-OH group, and one free water molecule. (Fig. 1a). The copper atom is tetracoordinate in an almost perfect square geometry with a CuO4 coordination environment formed by the three oxygen atoms (O1, O4#2 and O3#1 of three different bcpmba3− ligand and the one oxygen atom O5 of the µ4-OH group). The Cu–O bond lengths fall in the normal range of 1.91–1.95 Å [20]. The sum of the angles (∠O1–Cu1–O4#2 85.33(17)°, ∠O4#2–Cu1–O3#1 90.66(17)°, ∠O3#1–Cu1–O5 90.64(18)°, and ∠O5–Cu1–O1 93.37(17)°) is ~ 360°. The copper atom and four coordinated O atoms lie almost in the base plane, and the copper atom deviates from the base plane by 0.0041 Å. In compound 1, three carboxylate groups of bcpmba3− ligands are completely deprotonated and display a bridging bidentate coordination mode (Scheme 2a). Two adjacent copper atoms are bridged by sharing O5 to generate dimer units with a Cu1···Cu1 distance of 3.485 Å. Two Cu2O7 units are bridged by one oxygen atom of µ4-OH to generate a 1D chain along the c-direction (Fig. 1b). These chains are further bridged through four carboxylate O atoms from one bcpmba3− ligand to generate 2D porous layers in the ab-plane (Fig. 1c). The 2D porous layer is further extended by bcpmba3− linkers along the c-axis to generate a 3D porous framework. The solvent-accessible volume was calculated to be 27.5% using the PLATON software [19].

Crystal structure of 1: a Coordination environment of Cu(II) in 1 showing 50% probability displacement ellipsoids (H atoms are omitted for clarity). Symmetry code: #1 − x + 1/2, y + 1/2, − z + 3/2; #2 − x + 1/2, − y + 1/2, − z + 1; #3 − x + 1, y, − z + 5/2; #4 − x + 1/2, y − 1/2, − z + 3/2. b The 1D zigzag chain formed by Co atoms that are bridged by carboxylate O atoms. c The 2D layer in the ab plane of coordination polymer 1

The different coordination modes of 1 (a), 2 (b), 3(c)
[Mn(Hbcpmba)]n (2). Compound 2 crystallizes in the orthorhombic space group Pna21 and exhibits a 3D microporous framework. The asymmetric unit is composed of one manganese atom and one partly deprotonated Hbcpmba2− ligand. The coordination geometry around the manganese atom is a distorted octahedral, in which the four equatorial positions are occupied by the carboxylate oxygens (O4, O6#1, O2, and O4#2) with the bond lengths ranging from 2.152(4) to 2.294(4) Å, comparable to similar complexes reported in the literature [21]. Further, the axial positions are occupied by the carboxylate oxygens (O1 and O5) [Mn–O1 = 2.101(4) Å and Mn–O5 = 2.155(4) Å]. The bond angles around the Mn atom are in the range of 84.9–168.6° (∠O4–Mn1–O6#1 94.2(1)°, ∠O6#1–Mn1–O2 84.9(1)°, ∠O2–Mn1–O4#2 86.4(1)°, ∠O4#2–Mn1–O4 94.3(1)°, and ∠O5–Mn1–O1 168.6(2)°) (Fig. 2a). The manganese atoms are bridged as a dimer by sharing one oxygen atom to induce a 1D Mn–O–Mn zigzag chain with an Mn···Mn separation of 3.731(2) Å along the a-axis (Fig. 2b). However, the adjacent chains are linked by bcpmba3− ligands through monodentate, bridging bidentate, and bridging tridentate coordination modes to form 2D porous layers in the ab-plane (Scheme 2b). In this layer, each bcpmba3− ligand connects to four manganese atoms, and each manganese atom is connected to four ligands (Fig. 2c). These layers are further interconnected through bcpmba3− ligands, resulting in a 3D porous framework. The solvent-accessible volume was calculated to be 14.3% using the PLATON software [19].

Crystal structure of 2: a Coordination environment of Mn(II) in 2 showing 50% probability displacement ellipsoids (H atoms are omitted for clarity). Symmetry code: #1 x, y, z − 1; #2 − x + 1, − y + 1, z + 1/2; #3 − x + 1/2, y + 1/2, z − 3/2; #4 − x + 1, − y + 1, z − 1/2; #5 − x + 1/2, y − 1/2, z + 3/2; #6 x, y, z + 1; #7 x + 1/2, − y + 1/2, z − 1; #8 x − 1/2, − y + 1/2, z + 1. b The 1D zigzag chain formed by Mn atoms that are bridged by carboxylate O atoms. c The 2D layer in the ab plane of coordination polymer 2
[Co2(bcpmba)(μ3-OH)·H2O]n (3). Compound 3 crystallized in triclinic space group Pī. As indicated in Fig. 3a, 3 consists of two crystallographically independent cobalt atoms, one bcpmba3− ligand, one hydroxyl group, and a water ligand. The water molecule is highly disordered over two places with half occupancies. The cobalt atom is five coordinated by four carboxylate oxygen atoms from four bcpmba3− ligands, one oxygen atom from hydroxyl group to form triangular bicone coordination geometry, in which the equatorial positions are occupied by the three oxygen atoms (O2#1, O8#3 and O9) with the bond lengths ranging from 1.964(4) to 1.991(4) Å, comparable to similar complexes reported in the literature [22]. The bond angles around the cobalt 1 atoms are in the range of 101.01(18) to 173.91(14)°. The coordination geometry around cobalt 2 atoms is a distorted octahedral, in which the four equatorial positions are occupied by the oxygen atoms (O9#5, O9, O7#3 and O1W) with the bond lengths ranging from 2.069(4) to 2.099(4) Å, comparable to similar complexes reported in the literature [23]. Further, the axial positions are occupied by the carboxylate oxygens (O1#4 and O5) [Co2–O1#4 = 2.067(5) Å and Co2–O5 = 2.132(4) Å]. The bond angles around the cobalt atoms are in the range of 80.83(17) to 168.43(19)°. The deprotonated bcpmba3− anions adopt a bridging bidentate coordination mode to bridge four cobalt centers to form a tetranuclear cluster (Fig. 3b), then were connected with two carboxylic by bridging bidentate and monodentate coordination mode, extending to be the 1D chain (Fig. 3c). These chains are further linked by bcpmba3− ligands through bridging bidentate and bridging tridentate coordination modes to form 2D porous layers in the bc-plane (Fig. 3d). The 2D porous layer is further extended by bcpmba3− linkers along the a-axis to generate a 3D porous framework (Figure S3). The porosity was calculated using PLATON software at 19.2%.

Crystal structure of 3: a Coordination environment of Co(II) in 3 showing 50% probability displacement ellipsoids (H atoms are omitted for clarity). Symmetry code: #1 x + 1, y + 1, z; #2 − x + 2, − y + 1, − z + 1; #3 − x + 3, − y + 1, − z; #4 − x + 1, − y, − z + 1; #5 x + 1, y, z; #6 x − 1, y, z; #7 x − 1, y − 1, z. b The tetranuclear copper cluster. c The 1D zigzag chain formed by Co atoms that are bridged by carboxylate O atoms. d The 2D layer in the ab plane of coordination polymer 3
Effect of the coordination mode of ligand L on the structures of compounds 1–3
Notably, the coordination mode of ligands plays an important role in coordination chemistry [5, 24]. Coordination polymers 1–3 consist of 1D chains extended via the bridge of bcpmba3− ligands to generate 2D porous layers which are further connected by bcpmba3− ligands to generate 3D porous coordination polymers. However, it was observed that the pore size of coordination polymers is different, which can be attributed to different coordination modes of the ligand. The ligand adopts a Y-shaped configuration in all of the coordination polymers 1–3. However, coordination mode is different in the different coordination polymers. In coordination polymer 1, the three carboxylic groups of the ligand adopt bridging bidentate coordination mode. In coordination polymer 2, the ligand adopts monodentate, bridging bidentate, and bridging-tridentate coordination modes. For 3, the three carboxylic groups of the ligand adopt bridging bidentate and bridging-tridentate coordination mode (Scheme 2).
Different coordination modes of the ligands resulted in the difference in the pore size of the three coordination polymers. Therefore, the above-mentioned analyses revealed that the coordination modes of ligands often play an important role in the self-assembly processes to form porous coordination polymers with different structures.
PXRD and TGA analyses
Complexes 1–3 are air stable, insoluble in common organic solvents, and can retain their crystalline integrity under ambient conditions. In order to check the phase purity of complexes 1–3, the powder X-ray diffraction patterns of these complexes were checked at room temperature. As shown in Fig. S4–S6, the peak positions of the simulated and experimental PXRD patterns are in agreement with each other generally, demonstrating the good phase purity of the complexes. The thermal behaviors of 1–3 were studied by thermogravimetric analysis (TGA). The experiments were performed on samples consisting of numerous single crystals under a nitrogen atmosphere with a heating rate of 5 °C min−1, as shown in Fig. 4.

The thermogravimetric analyses (TGA) curves of coordination polymer 1–3
For 1, first, a gradual mass loss was discovered between 30 and 180 °C, which can be attributed to the departure of seven water molecules (found 18.6%; calcd 18.3%). The TGA trace of the framework then shows a plateau of stability until 280 °C. Further heating above 280 °C induced the gradual decomposition of 1. An abrupt weight loss was observed in the temperature range of 280–452 °C. For 2, the structure is stable up to 340 °C, with a total weight loss of 82.2% in the temperature range 340–488 °C, corresponding to the loss of Hbcpmba2− ligand (calcd 84.9%). The remaining residues corresponded to the formation of MnO (found 14.4%; calcd 14.9%). For complex 3, a weight loss of 15.0% (calcd 15.3%) is observed from 31 to 102 °C, which is attributed to the loss of the coordinated water and free water, next a weight loss of 59.1% (calcd 58.9%) is observed from 305 to 457 °C, which is attributed to the loss of the coordinated bcpmba 3− ligand.
Magnetic properties
Variable-temperature magnetic susceptibility studies were carried out on powder samples of 1–3 in the 2–300 K temperature range. As the temperature was lowered to 2 K, the χMT value continuously decreased, which suggests that antiferromagnetic interactions are operative in 1–3.
For compound 1, The χMT versus T plot exhibits a value of 0.38 cm3 K mol−1 at 300 K, close to the expected value of 0.37 cm3 mol−1 K for one spin-only copper atom (Fig. 5). The inverse susceptibility versus temperature plot is linear above 140 K, following the Curie–Weiss law with a Weiss constant of θ = − 155 K and a Curie constant of 0.58 cm3 mol−1 K (Figure S4). Further, the negative θ value indicates the presence of antiferromagnetic interactions among the adjacent copper atoms.

Temperature dependence of χMT (□) and χM (ο) versus T for coordination polymer 1
For compound 2, as shown in Fig. 6, the χMT value at 300 K is 4.43 cm3 mol−1 K, which is close to the value of 4.38 cm3 mol−1 K expected for one magnetically isolated high-spin Mn(II) center (SMn = 5/2, g = 2.0). Between 14 and 300 K, the magnetic susceptibility can be fitted to the Curie–Weiss law with C = 4.35 cm3 mol−1 K and θ = − 35.8 K (Figure S5). These results indicate an antiferromagnetic interaction between the adjacent Mn(II) ions. Compound 2 contains an Mn(II) chain interlinked by the H3bcpmba ligands. The exchange interaction is negligible because of a quite long distance (20.96 Å) between chains. Therefore, the system can be treated as a magnetically isolated 1D chain. In order to understand quantitatively the magnitude of magnetic interaction, a 1D chain model was used to simulate the experimental magnetic behavior. The interaction (J) can be extracted by the spin Hamiltonian H = − J ΣSi Si+1. In the classical-spin approximation, the following expression [Eq. (1)] of magnetic susceptibility was deduced by Fisher [25]
where μ is the Langevin function μ = coth [J S (S + 1)/kT] − kT/[J S (S + 1)], with S = 5/2. The best simulation of the experimental data of 2 leads to J = − 2.76 cm−1, g = 2.07 with an agreement factor [defined as R = Σ[(χM)obs − (χM)calc]2/Σ(χM)2obs] of 2.76 × 10−3. The solid lines in Fig. 6 show that the data in the 2–300 K can be explained using Eq. (1). The J parameter confirms that an antiferromagnetic exchange coupling exists between the adjacent Mn(II) centers, which is agreement with a negative θ value.

Temperature dependence of χMT (□)and χM (ο) versus T for coordination polymer 2. The solid line represents the best fit to the equation in the text
The temperature dependence of the magnetic susceptibility of 3 in the form of χMT and χM versus T is displayed in Fig. 7. The experimental χMT values at room temperature is 5.48 cm3 mol−1 K, which is greater than that for two isolated spin only Co2+ cations (3.75 cm3 mol−1 K with g = 2.0). The large value is due to the occurrence of an unquenched orbital contribution typical of the 4T1g ground state in six-coordinated Co (II) complexes [26]. The temperature dependence of the reciprocal susceptibilities (1/χM) obeys the Curie–Weiss law above 25 K with a Weiss constant of θ = − 10.8 K, Curie constant of C = 2.80 cm3 mol−1 K (Figure S6). The negative θ value also suggests that antiferromagnetic interactions are operative in 3.

Temperature dependence of χMT (□) and χM (ο versus T for coordination polymer 3
Conclusion
Three porous coordination polymers based on a semirigid tripodal carboxylate ligand were successfully developed under similar reaction conditions. These compounds exhibited intriguing coordination features with 3D porous coordination polymers. The structural differences indicate that different coordination modes of the ligand play an important role in the self-assembly processes to form coordination polymers with different structures. This work will further enrich the synthesis and design of CPs based on semirigid tripodal carboxylate ligands, and the extendable work will construct stabilized and functionalized materials through employing a variety of analogous ligands.
References
Zhang WH, Ren ZG, Lang JP (2016) Chem Soc Rev 45:4995–5019
Liu D, Lang JP, Abrahams BF (2011) J Am Chem Soc 133(29):11042–11045
Wan XY, Jiang FL, Chen L, Pan J, Zhou K, Su KZ, Pang JD, Lyu GX, Hong MC (2015) CrystEngComm 17:3829–3837
Liu PP, Wang CY, Zhang M, Song XQ (2017) Polyhedron 129:133–140
Yang ZX, Qian Y, Yu JW, Zhai L, Zhang WW, Ren XM (2018) RSC Adv 8:25489–25499
Yan YT, Liu J, Yang GP, Zhang F, Fan YK, Zhang WY, Wang YY (2018) CrystEngComm 20:477–486
Hu KQ, Zhu LZ, Wang CZ, Mei L, Liu YH, Gao ZQ, Chai ZF, Shi WQ (2016) Cryst Growth Des 16:4886–4896
Gu J, Liang X, Cai Y, Wu J, Shi Z, Kirillov A (2017) Dalton Trans 46:10908–10925
Zou RQ, Zhong RQ, Du M, Kiyobayashi T, Xu Q (2007) Chem Commun 24:2467–2469
An J, Geib SJ, Rosi NL (2010) J Am Chem Soc 132:38–39
Liu TF, Lü J, Guo Z, Proserpio DM, Cao R (2010) Cryst Growth Des 10:1489–1491
Lin ZJ, Liu TF, Xu B, Han LW, Huang YB, Cao R (2011) CrystEngComm 13:3321–3324
Huang WH, Yang GP, Chen J, Chen X, Zhang CP, Wang YY, Shi QZ (2013) Cryst Growth Des 13:66–73
Wei XH, Yang LY, Liao SY, Zhang M, Tian JL, Du PY, Gu W, Liu X (2014) Dalton Trans 43:5793–5800
SAINT, Version 6.02a; Bruker AXS Inc.: Madison, WI, 2002
Krause L, Herbst-Irmer R, Sheldrick GM, Stalke D (2015) J Appl Cryst 48:3–10
Sheldrick GM (2008) Acta Cryst A 64:112–122
Sheldrick GM (2015) Acta Cryst A 71:3–8
Sluis PVD, Spek AL (1990) Acta Cryst A 46:194–201
Senchyk GA, Lysenko AB, Krautscheid H, Rusanov EB, Chernega AN, Kramer KW, Liu SX, Decurtins S, Domasevitch KV (2013) Inorg Chem 52:863–872
Shao YL, Cui YH, Gu JZ, Kirillov AM, Wu J, Wang YW (2015) Rsc Adv 5:87484–87495
Ding R, Huang C, Lu J, Wang J, Song C, Wu J, Hou H, Fan Y (2015) Inorg Chem 54:1405–1413
Zhu Z, Bai YL, Zhang L, Sun D, Fang J, Zhu S (2014) Chem Commun 50:14674–14677
Deng DS, Liu LL, Ji BM, Yin GJ, Du CX (2012) Cryst Growth Des 12:5338–5348
Fisher ME (1964) Am J Phys 32:343–346
Han ML, Bai L, Tang P, Wu XQ, Wu YP, Zhao J, Li DS, Wang YY (2015) Dalton Trans 44:14673–14685
Acknowledgements
This work was supported by the start-up foundation of Sichuan University of Science and Engineering (No. 2015RC29).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
11243_2020_433_MOESM1_ESM.doc
CCDC 1013133, 1015472 and 1985640 for 1–3 contain the supplementary crystallographic data for the compounds reported in this article. These data can be obtained free of charge on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK [Fax: +44 1223 336 033, e-mail: deposit@ccdc.cam.ac.uk] or via www.ccdc.cam.ac.uk/data_request/cif. (DOC 1845 kb).
Rights and permissions
About this article

Cite this article
Sun, Y., Yin, M., Chen, S. et al. Synthesis, crystal structures and magnetic properties of three porous coordination polymers based on a semirigid tripodal carboxylate ligand. Transit Met Chem 46, 167–175 (2021). https://doi.org/10.1007/s11243-020-00433-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-020-00433-5