
Abstract
The Suzuki–Miyaura reaction of various aryl halides with aryl boronic acids using {[Ph2PCH2PPh2CH=C(O)(C10H7)]PdCl2} as a catalyst has been investigated. The X-ray crystal structure of the catalyst reveals a five-membered chelate ring formed by coordination of the ligand through the phosphine group and the ylidic carbon atom to the metal center. This palladacycle exhibited excellent activities and reusability in the aqueous phase for the Suzuki cross-coupling reactions of arylboronic acids with aryl halides. The proposed protocol featured mild reaction conditions and notable simplicity and efficiency using Cs2CO3 as a base in water. The catalytic system could be reused four times without significant loss of activity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The Suzuki–Miyaura coupling reaction has proven to be a very useful procedure for the synthesis of diverse biaryls, which constitute an important class of compounds for pharmaceutical, materials and agricultural chemistry [1–7]. Palladium catalysis is ubiquitous in modern organic chemistry, and a great number of catalytic systems have been designed and fine-tuned through careful ligand design and choice of experimental conditions. In Suzuki reactions, the activity of the catalyst depends on the nature of the ligand attached to the Pd metal [8]. Bulky electron-rich phosphine ligands are prominent in the palladium-catalyzed Suzuki cross-coupling reaction, due to their superior donor capability and stabilization effects [9–12]. In recent years, various palladium–phosphine catalysts have been developed for efficient cross-coupling reactions [13–20], but most of the known systems give sub-optimal results for the Suzuki coupling of less reactive aryl halides. Furthermore, a high loading of catalyst and an inert atmosphere in most reactions, especially those involving phospha–palladacycles, are generally required to achieve high conversions [21]. Accordingly, this field is still challenging, and novel developments and greener protocols for the Suzuki cross-coupling reaction are desirable.
In 1981, the application of Pd(PPh3)4 in a Suzuki coupling reaction was reported for the first time [22]. Recently, palladacycle complexes have been employed in Suzuki reactions, as active and more air-inert catalytic candidates [23–26]. In this context, we were interested to explore the use of a five-membered chelate ring palladacycle, whose X-ray crystal structure has been reported in our previous work [27], as a catalyst precursor for Suzuki cross-coupling in aqueous environments (Scheme 1). This complex gives a high catalytic activity in the coupling reactions of various aryl halides and aryl boronic acids under aerobic conditions. Easy catalyst recovery and excellent recycling efficiency of the catalyst make it an ideal system for coupling reactions in water as solvent. The results of our investigations are reported in this paper.

Synthetic route for preparation of palladacycle complex
Experimental
All chemicals obtained from commercial suppliers were reagent grade and used without further purification. Fourier transform IR spectra were recorded on a Shimadzu 435-U-04 spectrophotometer as KBr pellets, in the 200–4000 cm−1 region. NMR spectra were obtained on 400 and 250 MHz Bruker spectrometers in CDCl3 or DMSO-d6 as solvent. Melting points were determined using an SMP3 apparatus.
Cyclic voltammetry was performed using an Autolab model PGSTAT 20 potentiostat/galvanostat. The working electrode used in the voltammetry experiments was a Pt disk (1.8 mm diameter), and a platinum wire was used as the counter electrode. The working electrode potentials were measured versus the Ag/AgCl electrode (all electrodes were purchased from AZAR Electrodes co.).
The X-ray diffraction measurements were made on a STOE IPDS-II diffractometer with graphite-monochromated Mo Kα radiation. A yellow crystal of the complex with dimensions of 0.21 × 0.20 × 0.11 mm was mounted on a glass fiber and used for data collection. Cell constants and an orientation matrix for data collection were obtained by least-squares refinement of the diffraction data. Data were collected at a temperature of 298(2) K in a series of ω-scans in 1° oscillations and integrated using the Stoe X-AREA [28] software package. A numerical absorption correction was applied using X-RED [29] and X-SHAPE [30] software. The structure was solved by direct methods [31] and subsequent difference Fourier maps and then refined on F 2 by full-matrix least-squares procedures using anisotropic displacement parameters [31]. All of the hydrogen atoms were located in a difference Fourier map and refined isotropically. Subsequent refinement was then converged with R factors and parameter errors that became significantly better for all attempts to model the solvent disorder. All refinements were performed using the X-STEP32 crystallographic software package [32]. During the structure analysis, it was observed that the unit cell contains large accessible voids, which host disordered solvent molecules. Since the solvent molecules could not be located, the PLATON/SQUEEZE procedure was used [33].
General procedure for the Suzuki coupling reaction
A reaction tube was charged with the required aryl halide (0.75 mmol), aryl boronic acid (1 mmol), Pd catalyst (0.001 mmol) and Cs2CO3 (1.5 mmol) in water (2 mL). The reaction mixture was stirred for the required period of time at 80 °C until completion of the reaction, as monitored by TLC. The final reaction mixture was cooled to room temperature and extracted with n-hexane. The combined organic phase was dried with CaCl2, solvent was removed, and the product was recrystallized from ethanol. Yields were calculated against consumption of the aryl halides, and pure products were identified by FTIR, 1H and 13C NMR spectroscopy and melting point analysis (See supplementary data).
Results and discussion
The palladium(II) complex was prepared based on the previously published method [27]. The complex was recrystallized by slow evaporation over several days from dimethyl sulfoxide solution. The molecular structure is shown in Fig. 1. Relevant parameters concerning data collection and refinement are given in Table 1, and selected bond distances and angles are presented in Table 2.

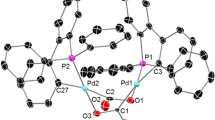
Ortep view of X-ray crystal structure of {[Ph2PCH2PPh2CH=C(O)(C10H7)]PdCl2}, in 50 % probability level
X-ray analysis of the complex reveals that the Pd atom is coordinated by the P atom of the phosphine group, the ylidic carbon and two chloride ligands, leading to a slightly distorted square-planar geometry around the metal. Hence, the Ph2PCH2PPh2CH=C(O)C10H7 ligand adopts a P,C-chelated mode. The Pd(1)–P(1) bond length in this complex [2.222(3) Å] is comparable to analogous distances in palladium complexes such as [PdCl2(dppe)] (dppe = 1,2-(diphenylphosphino)ethane) [2.233(2) and 2.226(2) Å] [34] and {[(Ph2PCH2PPh2CH=C(O)C6H4Ph)]PdCl2} [2.2289 Å] [27]. The Pd–C(ylide) bond length in this complex [2.100(11) Å] is similar to those found in [PdCl2{PPh2C(H)C(O)CH2PPh2C(H)}] [2.099(6) and 2.104(6) Å] [35]. The stabilized resonance structure of the parent ylide is lost upon the complex formation; thus, the C(26)–C(27) bond length in the orthopalladated complex [1.505(17) Å] is significantly longer than the corresponding distance found in the uncomplexed parent ylides such as Ph2PCH2PPh2CH=C(O)C6H4Ph [1.253(3) Å] [35]. The bond length of P–C(H) in the parent ylides such as Ph2PCH2PPh2CH=C(O)C6H4Ph is 1.722(3) Å [36], while the corresponding bonds are considerably elongated to 1.776(11) Å in this complex. The angles subtended by the ligand at the Pd(II) center vary from 89.25(12)° to 91.88(12)° (the sum of the angles is 360.83°) and from 172.66(12)° to 173.8(3)°, revealing the distortion from a square-planar environment. An interesting structural feature concerns the crystal packing, where intermolecular interactions are observed. Thus, the packing is stabilized by intermolecular H bonds of the type C–H…Cl (Cl1–H50A = 2.724 Å, Cl1–H62 = 2.861 Å and Cl2–H47 = 2.787 Å), as shown in Fig. 2.

Best view of the palladacycle complex revealing the supramolecular packing. Intermolecular H bonds of the type C–H…Cl are shown as dashed lines
The electrochemical behaviors of the free ylide, Y, and its Pd(II) complex were studied by cyclic voltammetry. Figure 3 shows the cyclic voltammogram obtained for 1 mM Y in acetonitrile containing 0.1 M tetra-n-butylammonium perchlorate (Bu4NClO4) as supporting electrolyte, recorded with a platinum electrode, which shows that reduction of Y occurs at the first stage. Under these conditions, the voltammogram exhibits a reduction peak CL1 and two anodic peaks (AL1 and AL2).

Cyclic voltammogram of 1 mM of Y in acetonitrile, containing 0.1 M tetra-n-butylammonium perchlorate (Bu4NClO4) as supporting electrolyte, at Pt electrode. Sweeping direction: reduction of Y at the first stage and oxidation at the second stage. Scan rate: 100 mV s−1; t = 25 °C. Vectors show sweeping direction
The cyclic voltammogram of a 1 mM solution of the palladacycle complex, under the same conditions, showed irreversible electron transfer. At a scan rate of 100 mV/s, there were two cathodic peaks; CL1, corresponding to the reduction of Y, and CM1 at 1.05 V versus the Ag/AgCl reference electrode, corresponding to the redox pair Pd(II)/Pd(0). A single anodic peak (AL1) was observed on the reverse scan. This behavior is similar to that reported previously by Champness et al. [37], Batchelor et al. [38], Downard et al. [39] and our group [40–42] in cyclic voltammetric studies of various types of Pd(II) complexes in aprotic solvents. The irreversibility may be due to the reaction of a Pd(0) complex with adventitious O2 or other components of the solution [39].
The cathodic peak CM1 at 1.05 V versus the Ag/AgCl reference electrode can be assigned to the formation of a Pd(0) center with a coordination number between 2 and 4 [or alternatively to an equilibrium mixture of several Pd(0) species] [39, 43–45]. There are some examples of stable Pd(0) complexes; particularly relevant is [Pd(L)2]0 (L = 1,2-bis(dimethylarsino)benzene), which is sufficiently stable to be isolated as a solid [46]. These electrochemical data confirm complex formation between Pd(II) and Y and also show the presence of such Pd(II) in the complex. The overall two-electron reduction of the Pd(II) complexes is expected to take place in separate one-electron steps [37–39], and this might be achieved via manipulation of the thermodynamics or electrode kinetics of either step by correct choice of the experimental conditions. Under our experimental conditions, the Pd(I) complex is unstable. Thus, peak CM1 in Fig. 4 corresponds to a two-electron reduction of the Pd(II) complex which is a consequence of the thermodynamic instability of the intermediate one-electron product under our experimental conditions.

Cyclic voltammogram of 1 mM of the Pd(II) complex in acetonitrile, containing 0.1 M tetra-n-butylammonium perchlorate (Bu4NClO4) as supporting electrolyte, at Pt electrode. Sweeping direction: reduction of complex at the first stage and oxidation at the second stage. Scan rate: 100 mV s−1; t = 25 °C. Vectors show sweeping direction
The catalytic activity of the palladacycle complex was assessed in the Suzuki cross-coupling reaction, initially by studying the coupling of iodobenzene with phenylboronic acid to give biphenyl as the sole product [47]. Various parameters including solvent, base and catalyst loading were screened to optimize the reaction conditions. Initially, the effect of catalyst loading was investigated, using different quantities of the catalyst ranging from 0.0005 to 0.10 mmol. The best results were obtained using 0.001 mmol of catalyst (Table 3). No significant improvement in the reaction results was observed upon further increasing the quantity of catalyst. A series of bases including Cs2CO3, K2CO3, Na2CO3, NaOAc and Et3N were then screened, with the optimum results obtained using aqueous Cs2CO3 (up to 93 % isolated yield at 80 °C; Table 4). Additionally, to study the effect of solvent on the reaction, we examined a range of solvents in comparison with water (e.g., EtOH, DMF, NMP and toluene). The best results obtained with alternative solvents to water did not provide any improved yields under the conditions employed (Table 4).

Using the optimized conditions (solvent: H2O, base: Cs2CO3; quantity of catalyst: 0.001 mmol, temperature: 80 °C), the scope of the reaction was subsequently investigated for several substrates. The results summarized in Table 5 clearly demonstrate that the protocol was amenable to various substrates and substituents, providing very good to excellent yields even in the case of less activated (chloro-derived) substrates, with relatively short reaction times in the range of 0.5–2 h. Various functionalized aryl halides incorporating –NO2, –CH3, –CHO, –OCH3 and –COCH3 substituents reacted with aryl boronic acids to give the corresponding coupling products. Better yields were achieved for aryl halides with electron-withdrawing rather than electron-donating substituents. Although the electronic properties of aryl iodides did not significantly affect the catalyst performance, these reactions proceeded very rapidly, resulting in excellent yields in 20–45 min for both electron-deficient and electron-rich aryl iodides (Table 5, entries 1 and 2). Cross-coupling reactions of aryl boronic acids with aryl bromides incorporating electron-donating substituents such as –CH3 and –OCH3 groups and electron-withdrawing substituents such as –NO2, –COCH3 and –CHO groups, and 3-bromopyridine were transformed efficiently to the biaryl products. The reaction of electron-neutral bromobenzene with boronic acid derivatives gave excellent yields (Table 5, entry 3). For electron-rich or deactivated p-bromotoluene and p-bromoanisole, the reaction gave yields around 80 %, indicating that the coupling was sensitive to the electron density on the aryl bromides due to the methyl and methoxy groups, respectively (Table 5, entries 4 and 6). As expected, couplings of the electron-poor or activated 4-bromonitrobenzene, 4-bromoacetophenone, 4-bromobenzaldehyde and 3-bromopyridine with aryl boronic acids generally gave excellent yields (Table 5, entries 5, 7, 8 and 9). To extend the scope of our work, we next investigated the coupling of electron-rich and electron-deficient aryl chloride substrates with organoboron compounds. While aryl bromides or iodides reacted readily, aryl chlorides showed slower reaction rates due to the stronger C–Cl bond. Aryl chlorides with electron-withdrawing groups (Table 5, entry 11) reacted more easily than those with electron-donating groups (Table 5, entry 10).
In order to investigate the recyclability of the catalyst, the Suzuki cross-coupling reaction of iodobenzene with phenylboronic acid and ethyl phenylboronic acid catalyzed by 0.001 mmol of the Pd catalyst was chosen as a model reaction. After completion of the reaction, the mixture was filtered, and the recovered catalyst was washed with n-hexane and dried under reduced pressure.
The results depicted in Table 6 confirm that the Pd catalyst could be recycled and reused several times without any appreciable loss of activity.
Conclusion
In summary, we successfully developed a palladium–phosphine complex, as a highly active and stable catalyst for Suzuki cross-coupling reaction in aqueous phase under mild conditions. The ease of synthesis of the complex together with its high efficiency, very low catalyst loading, high yields and easy recyclability makes it an ideal catalytic system for coupling reactions. Further investigations on the application of this class of catalyst are currently in progress in our laboratory.
Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://www.editorialmanager.com/tmch/download.aspx?id=72101&guid=50b09e7e-fd16-4e27-9c70-2b8189e841fd&scheme=1 CCDC 987318 contains the supplementary crystallographic data for the complex. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif or from the Cambridge Crystallographic Data Center, 12, Union Road, Cambridge CB2 1EZ, UK. Tel.: +44 0 1223 762911; or deposit@ccdc.cam.ac.uk.
References
Zhou Z-Z, Liu F-S, Shen D-S, Tan C, Luo L-Y (2011) Inorg Chem Commun 14:659–662
Littke AF, Fu GC (2002) Angew Chem Int Ed 41:4176–4211
Alonso F, Beletskaya IP, Yus M (2008) Tetrahedron 64:3047–3101
Polshettiwar V, Decottignies A, Len C, Fihri A (2010) ChemSusChem 3:502–522
Teo S, Weng Z, Hor T (2011) J Organomet Chem 696:2928–2934
Tang Y-Q, Lu J-M, Shao L-X (2011) J Organomet Chem 696:3741–3744
Lasri J, MacLeod TC, Pombeiro AJ (2011) Appl Catal A Gen 397:94–102
Alizadeh A, Khodaei M, Kordestani D, Beygzadeh M (2013) Tetrahedron Lett 54:291–294
Weng Z, Teo S, Hor TA (2007) Acc Chem Res 40:676–684
Fu GC (2008) Acc Chem Res 41:1555–1564
Molander GA, Canturk B (2009) Angew Chem Int Ed 48:9240–9261
Martin R, Buchwald SL (2008) Acc Chem Res 41:1461–1473
Punji B, Ganesamoorthy C, Balakrishna MS (2006) J Mol Catal A Chem 259:78–83
Bedford RB, Welch SL (2001) Chem Commun, pp 129–130
Aizawa S-I, Hase T, Wada T (2007) J Organomet Chem 692:813–818
Chahen L, Therrien B, Süss-Fink G (2006) J Organomet Chem 691:4257–4264
Milde B, Schaarschmidt D, Ecorchard P, Lang H (2012) J Organomet Chem 706:52–65
Aizawa S-I, Majumder A, Yokoyama Y, Tamai M, Maeda D, Kitamura A (2009) Organometallics 28:6067–6072
Milde B, Packheiser R, Hildebrandt S, Schaarschmidt D, Rüffer T, Lang H (2012) Organometallics 31:3661–3671
Sabounchei SJ, Panahimehr M, Ahmadi M, Akhlaghi F, Boscovic C (2014) C R Chim 17:81–90
Frey GD, Schütz J, Herdtweck E, Herrmann WA (2005) Organometallics 24:4416–4426
Miyaura N, Yanagi T, Suzuki A (1981) Synth Commun 11:513–519
Farina V (2004) Adv Synth Catal 346:1553–1582
Dupont J, Consorti CS, Spencer J (2005) Chem Rev 105:2527–2572
Subhas MS, Racharlawar SS, Sridhar B, Kennady PK, Likhar PR, Kantam ML, Bhargava SK (2010) Org Biomol Chem 8:3001–3006
Sabounchei SJ, Ahmadi M, Panahimehr M, Bagherjeri FA, Nasri Z (2014) J Mol Catal A Chem 383:249–259
Sabounchei SJ, Panahimehr M, Ahmadi M, Nasri Z, Khavasi HR (2013) J Organomet Chem 723:207–213
D. S. C. GmbH, 1.30 ed., Germany, 2005, Program for the acquisition and analysis of data
D. S. C. GmbH, Germany., 1.28b ed., 2005, Program for data reduction and absorption correction
D. S. C. GmbH, Germany., 2.05 ed., 2004, Program for crystal optimization for numerical absorption correction
Sheldrick GM (2007) Acta Crystallogr A 64:112–122
D. S. C. GmbH, Germany., 1.07b ed., 2000, Crystallographic package
Spek AL (2009) Acta Crystallogr D65:148–155
Steffen W, Palenik GJ (1976) Inorg Chem 15:2432–2439
Falvello LR, Margalejo ME, Navarro R, Urriolabeitia EP (2003) Inorg Chim Acta 347:75–85
Sabounchei SJ, Panahimehr M, Khavasi HR, Bagherjeri FA, Boscovic C (2014) Chem Pap 68:624–632
Champness NR, Kelly PF, Levason W, Reid G, Slawin AM, Williams DJ (1995) Inorg Chem 34:651–657
Batchelor RJ, Einstein FW, Gay ID, Gu J, Pinto BM, Zhou X (1996) Inorg Chem 35:3667–3674
Downard AJ, Bond AM, Clayton AJ, Hanton LR, McMorran DA (1996) Inorg Chem 35:7684–7690
Sabounchei SJ, Samiee S, Nematollahi D, Naghipour A, Morales-Morales D (2010) Inorg Chim Acta 363:3973–3980
Sabounchei SJ, Shahriary P, Salehzadeh S, Gholiee Y, Nematollahi D, Chehregani A, Amani A (2014) New J Chem 38:1199–1210
Sabounchei SJ, Shahriary P, Salehzadeh S, Gholiee Y, Nematollahi D, Chehregani A, Amani A, Afsartala Z (2014) Spectrochim Acta A 135:1019–1031
Cotton FA (1998) A comprehensive text. Intersci, p 917
Wilkinson G, Gillard R, McCleverty J (1987) Pergamon, Oxford, pp 534–774
Sabounchei SJ, Ahmadi M, Nasri Z, Shams E, Salehzadeh S, Gholiee Y, Karamian R, Asadbegy M, Samiee S (2013) C R Chim 16:159–175
Chatt J, Hart F, Watson H (1962) J Chem Soc, pp 2537–2545
Marziale AN, Jantke D, Faul SH, Reiner T, Herdtweck E, Eppinger J (2011) Green Chem 13:169–177
Acknowledgments
We are grateful to Bu-Ali Sina University for financial support.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Sabounchei, S.J., Hosseinzadeh, M., Panahimehr, M. et al. A palladium–phosphine catalytic system as an active and recycable precatalyst for Suzuki coupling in water. Transition Met Chem 40, 657–663 (2015). https://doi.org/10.1007/s11243-015-9959-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-015-9959-5