
Abstract
This work presents two complexes with general formula [RuCl3(NO)(PHOX)] (PHOX = 2-[2-(diphenylphosphino)phenyl]-4,4-dimethyl-2-oxazoline). Reaction of the PHOX ligand with [RuCl3(H2O)2(NO)] in refluxing methanol afforded fac-[RuCl3(NO)(PHOX)] (1), whereas exposure of a dichloromethane solution of complex 1 to ambient light gave mer,trans-[RuCl3(NO)(PHOX)] (2). On the other hand, reflux of complex 2 in methanol for 3 h resulted in thermal isomerization, to furnish complex 1. 1H and 31P NMR, elemental analysis, vibrational spectroscopy, UV–Vis, cyclic voltammetry, and X-ray diffractometry (for complex 2) aided characterization of the complexes. This work also compares the chemical properties of complexes 1 and 2 with the properties of similar complexes bearing PMA ([o-(N,N-dimethylamino)phenyl]diphenylphosphine, DPPE (1,2-bis(diphenylphosphino)ethane), and DPPP (1,3-bis(diphenylphosphino)propane); it also offers some insight into photo- and thermal isomerization mechanisms.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Potentially chelating ligands bearing phosphorus and nitrogen as donor atoms (P–N) have been the object of extensive studies in the field of coordination chemistry. Their coordination to a metal center affords highly stable complexes. Investigations into P–N ligand complexes have focused on catalysis mainly, because the presence of two atoms with different characteristics in the same complex may result in hemilabile properties and give rise to a vacant coordination site [1–3].
Work on the chemistry of ruthenium complexes displaying P–N ligands started in 1975, when Rauchfuss synthesized the complex [RuCl2(PMA)2] (PMA = [o-(N,N-dimethylamino)phenyl]diphenylphosphine, Fig. 1a) [3]. Since then, several papers have reported on ruthenium complexes containing this ligand [4–9], which has made PMA one of the most explored P–N ligands in ruthenium coordination chemistry. Examples of ruthenium complexes with imine-containing P–N ligands derived from pyridine, imidazole, and oxazoline (see Fig. 1b–d, respectively) are well documented [8, 10–16]. Oxazoline-containing P–N ligands, generally designated PHOX (phosphine oxazolines), are noteworthy and have been an extensively investigated subject matter in the past 30 years, especially in asymmetric catalysis [14–17].

Since 2006, we have devoted our attention to the reactivity of potentially hemilabile P–N and P–O ligands in complexes with carbonyl- or nitrosyl-containing ruthenium precursors [7–9, 22]. We have also isolated the first ruthenium nitrosyl complexes containing P–N [7, 9, 22].
Several complexes involving the “RuCl3(NO)” unit with arsines [23–25] and phosphines [23, 25–28] exist, but the literature provides only a few examples of complexes formed between the “RuCl3(NO)” unit and P–N ligands [7, 9, 22]. To expand our knowledge about the chemistry of ruthenium nitrosyl complexes with P–N ligands, this work describes the synthesis, characterization, and properties of the complexes fac- (1) and mer,trans-[RuCl3(NO)(PHOX)] (2) (PHOX = 2-[2-(diphenylphosphino)phenyl]-4,4-dimethyl-2-oxazoline).
Experimental section and methods
Measurements
IR spectra were recorded on a FTIR Bomem–Michelson 102 spectrometer in the 4000–400 cm−1 region. To this end, solid samples were pressed in KBr pellets or dissolved in dichloromethane solution in a CaF2 crystal with path length of 1 mm. 31P{1H} and 1H NMR spectra were acquired on a Bruker AVANCE 200 NMR spectrometer equipped with a 5-mm multinuclear direct detection probe, at room temperature and 4.7 T, in CD2Cl2. 31P and 1H NMR chemical shifts are given in ppm relative to H3PO4 85 % and TMS (tetramethylsilane), respectively. Coupling constants are given in Hz, and the splitting of the hydrogen and phosphorus signals is defined as s = singlet, d = doublet, and m = multiplet. Cyclic voltammetry (CV) experiments were carried out on a PARC 273 (Princeton Applied Research) instrument, at room temperature, in CH2Cl2 or CH3CN containing 0.1 M [Bu4N]ClO4 (TBAP) (Fluka, purum). Under these conditions, the half-wave potential for ferrocene was 0.423 V. The working and auxiliary electrodes were stationary Pt foils; the reference electrode was Ag/AgCl in a Luggin capillary probe filled with the electrolyte solution (TBAP in CH2Cl2 or CH3CN). Electronic spectra were obtained on a Hewlett–Packard diode array 8452A spectrophotometer from dichloromethane solutions of the complexes placed in quartz cuvettes with a path length of 1 cm, at concentrations ranging from 10−6 to 10−2 mol L−1. Elemental analyses were performed on a Fisons CHNS-O, EA 1108 element analyzer.
X-ray diffraction data
Data were collected on a Bruker APEX II CCD area-detector diffractometer with graphite-monochromatic Mo-Kα (0.71073 Å) radiation. SHELXS helped to solve the structures by direct methods [29]. Subsequent Fourier-difference map analyses provided the positions of the non-hydrogen atoms. The SHELXL package aided accomplishment of refinements [29]. Full-matrix least squares on F 2 with anisotropic displacement parameters enabled refinements for all the non-hydrogen atoms. Hydrogen atoms were included in the refinement, at the calculated positions. Table 1 lists crystal data and more details on data collection and refinement.
Materials and methods
Commercially available RuCl3·3H2O was donated by Johnson Matthey plc and was used as received. [RuCl3(H2O)2(NO)] and 2-[2-(diphenylphosphino)phenyl]-4,4-dimethyl-2-oxazoline (PHOX) were prepared according to the method described in the literature [30, 31]. NO was generated by reaction of dilute nitric acid (ca 33 %) over copper metal. The NO gas was passed through a trap containing saturated NaOH solution. The gas was dried by passage through a column containing anhydrous CaC12. The solvents were dried prior to use. All the manipulations involving solutions of the complexes and ligands were performed under argon atmosphere.
Synthesis of complexes 1 and 2
The designations fac and mer refer to the relative positions of the chloro ligands; the designations trans and cis refer to the positions of the nitrosyl and the phosphorus atoms relative to each other.
fac-[RuCl3(NO)(PHOX)] (1)
PHOX (217 mg; 0.604 mmol) was added to a degassed methanol solution of [RuCl3(H2O)2(NO)] (118 mg; 0.432 mmol). The resulting suspension was refluxed for 3 h, to give an orange solid. After cooling of the suspension to room temperature, the orange solid was filtered off, washed with methanol (3 × 2 mL), and dried under vacuum. Yield (232 mg; 90 %). Anal. Calcd. for C23H22Cl3N2O2PRu: exp.(calc.) C–46.6 (46.3), H–3.8 (3.7), N–4.8 (4.7). IR: νNO 1872 cm−1 (KBr), νNO 1872 cm−1 (DCM). UV–Vis (CH2Cl2), λ/nm (ε/M−1 cm−1): 274 (1.05 x105), 365 (498), 453 (<100). 31P{1H} NMR (81 MHz, CD2Cl2): 29.3 ppm (s). 1H NMR (200 MHz, CD2Cl2): δ/(ppm) 8.3–6.9 (m, 14 H, aromatic hydrogen nuclei), 4.5 (d, 2 J H–H = 8.5 Hz, 1 H O–CH 2), 4.0 (d, 2 J H–H = 8.5 Hz, 1 H, O–CH 2), 1.9 (s, 3 H, CH 3), and 1.6 (s, 3 H, CH 3).
mer,trans-[RuCl3(NO)(PHOX)] (2)
Complex (1) (100 mg; 0.168 mmol) was dissolved in CH2Cl2, under argon, and the resulting orange solution was stirred for 3 days in the presence of ambient light. The resulting green solution was concentrated to ~1 mL, which was followed by addition of diethyl ether, to yield a green solid. After filtration, the solid was washed with diethyl ether and dried under vacuum. Suitable single crystals were obtained from slow evaporation of the dichloromethane solution. Yield (98 mg, 98 %). IR: νNO 1860 cm−1 (KBr), νNO 1865 cm−1 (DCM). UV–Vis (CH2Cl2): λ/nm (ε/M−1 cm−1) 360 (4.6 × 103), 400 (27), 424 (320), 615 (<100). 31P{1H} NMR (81 MHz, CD2Cl2): 20.6 pm (s). 1H NMR (200 MHz, CD2Cl2): δ/(ppm) 8.2–6.8 (m, 14 H, aromatic hydrogen nuclei), 4.3 (s, 2 H, O–CH 2), 1.4 (s, 6 H, {CH3}2).
Thermal isomerization (mer,trans(2) → fac (1))
A degassed methanol suspension of mer,trans-[RuCl3(NO)(PHOX)] (2) (50 mg, 0.084 mmol) was refluxed for 3 h, in the absence of light. The resulting orange suspension was filtered; the orange solid was washed with MeOH (2 × 5 mL) and dried under vacuum. The spectroscopic data were consistent with those obtained for complex (1). Yield (48 mg, 96 %).
Results and discussion
Synthesis and basic characterization
Reaction of [RuCl3(H2O)2(NO)] with the ligand 2-[2-(diphenylphosphino)phenyl]-4,4-dimethyl-2-oxazoline (PHOX) in refluxing methanol gave the complex fac-[RuCl3(NO)(PHOX)] (1). An orange solid separated after cooling of the solution to room temperature. Exposure of a dichloromethane solution of complex 1 to ambient light afforded the complex mer,trans-[RuCl3(NO)(PHOX)] (2) (note: the solution of complex 1 was stable for several days when kept in the absence of light). The orange solution of complex 1 started to change to green after a few hours. After 3 days, complex 2 separated as a green solid (see Scheme 1). The last section of this document will discuss photo- and thermal isomerization.

Synthetic route for the preparation of complexes 1 and 2
The 31P{1H} NMR spectra of complexes 1 and 2 displayed one singlet centered at 29.3 and 20.6 ppm, respectively. The shielding observed for complex 2 as compared with complex 1 stemmed from the structural trans effect (STE) [32] of the nitrosyl ligand on the Ru–P bond in complex 2. In general, the Ru–P bond length and the 31P NMR chemical shift are inversely related [13, 33]. The chemical shift of 31P is very sensitive to the nature of the ligand in the trans position. For example, the presence of ligands with strong structural trans effect, such as NO+ and CO, weakens the Ru–P bond [13, 33], shields the phosphorus nucleus, and decreases the chemical shift [28, 34, 35].
Comparison of the δ values of complexes 1 and 2 with the δ values of fac- and mer,trans-[RuCl3(NO)(PMA)] (35.6 and 28.3 ppm, respectively, [7, 9] ) clearly showed that the size of the chelating agent ring affected deshielding of the P–N ligand upon coordination. The Δδ (δ coord−δ free) values for complex 1, complex 2, fac-[RuCl3(NO)(PMA)], and mer,trans-[RuCl3(NO)(PMA)] were 34.8, 26.1, 49.6, and 42.3 ppm, respectively. Deshielding was higher for the chelating agent with the five-membered ring than for the chelating agent with the six-membered ring [36, 37]. fac- and mer-[RuCl3(NO)(DPPE)] and fac- and mer-[RuCl3(NO)(DPPP)] (DPPE = 1,2-bis(diphenylphosphino)ethane; DPPP = 1,3-bis(diphenylphosphino)propane) behave in the same way: Δδ values for complexes bearing DPPE (chelate ring size = 5) and DPPP (chelate ring size = 6) range from 47.3 to 58.6 ppm and from 18.5 to 33.3 ppm, respectively [28].
The 1H NMR spectrum of complex 1 displayed two singlets [at 1.59 ppm, 3 H, and at 1.88 ppm, 3 H] for the (CH 3)2 hydrogen nuclei and two doublets [at 3.98 ppm, 1 H, 2 J H–H = 8.54 Hz and at 4.49 ppm, 1H, 2 J H–H = 8.54 Hz] for the CH 2 -O hydrogen nuclei of the oxazoline ring. The non-equivalence between these hydrogen nuclei allowed us to conclude that complex 1 corresponded to the fac isomer. For complex 2, the 1H NMR spectrum revealed one singlet at 1.37 ppm, 6 H, for the two CH 3 and one singlet at 4.28 ppm, 2 H, for CH 2–O. This was compatible with meridional arrangement of the chloro ligands and agreed with the molecular structure obtained by single-crystal X-ray diffractometry (see Fig. 2). The deshielding observed for the methyl and carbinolic hydrogen nuclei as compared with the free ligand [δ = 1.75 ppm (s, 6 H) and 3.73 (s, 2 H), respectively] and the chemical shifts in the 31P{1H} NMR spectra support a chelated coordination mode for the PHOX ligand. In addition, the 1H NMR of both isomers revealed signals in the region between 6.84 and 8.27 ppm (14 H, m) for the aromatic hydrogen nuclei of the PHOX ligand.

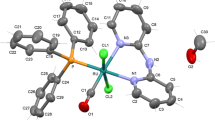
Representation of the structure of mer-[RuCl3(NO)(PHOX)] (2) showing the atoms labeling scheme. Atomic displacement ellipsoids are shown at 30 % probability level. Bond lengths (Å): Ru–N1 1.755(3), Ru–N2 2.105(2), Ru–P1 2.4626(8), Ru–Cl1 2.3879(9), Ru–Cl2 2.3473(9), Ru–Cl3 2.3612(9), N1–O1 1.133(4). Bond angles (°): N1–Ru–N2 97.89(11), N1–Ru–Cl1 84.62(10), N1–Ru–Cl2 96.73(10), N1–Ru–Cl3 90.96(9) N2–Ru–Cl1 91.03(8), N1–Ru–P1 170.76(10), N2–Ru–P1 80.08(7), Cl1–Ru–P1 86.39(3), Cl2–Ru–P1 92.39(3), Cl3–Ru–P1 90.94(3), Cl1–Ru–Cl2 175.48(3), Cl1–Ru–Cl3 88.27(3), Cl3–Ru–Cl2 87.40(3), O–N1–Ru 168.4(3)
Concerning the IR spectra, the νNO band emerged at 1872 cm−1 (KBr pellet) and 1871 cm−1 (CH2Cl2 solution) for complex 1, and at 1860 cm−1 (KBr pellet) and 1865 cm−1 (CH2Cl2 solution) for complex 2. These values lay in the range observed for other nitrosyl ruthenium complexes and are characteristic of RuII-NO+ species [28, 38–40].
X-ray structure
Slow evaporation of a dichloromethane solution of complex 2 afforded single crystals. Figure 2 illustrates the molecular structure of complex 2; the figure caption summarizes the relevant bond lengths and angles.
The geometry around ruthenium in complex 2 is best described as pseudo-octahedral, as evidenced by the bond lengths and angles (see caption of Fig. 2). The three chloro ligands occupy meridional positions, and the nitrosyl ligand is trans to the P atom.
Nitrosyl exerts a pronounced structural trans effect (STE) due to its strong π-accepting ability [32]. Consequently, the Ru–P bond length in complex 2 (2.4626(8) Å) is longer than the Ru–P bond in fac-[RuCl3(NO)(PMA)] (2.3241(6) Å) [9] and in other ruthenium nitrosyl analogues bearing phosphorus trans to a chloro ligand [28]. When NO+ is trans to a π-acceptor ligand, both compete for the dπ electrons of ruthenium, and NO+ weakens the Ru–L trans bond [32]. The Ru–P bond length in complex 2 is longer than the Ru–P bond length in mer,trans-[RuCl3(NO)(PMA)] (2.4038(9) Å) [7]—probably, the fact that PMA is a more basic N-donor (sp3) than PHOX (sp2) makes the ruthenium center richer in electron density and favors Ru → P back bonding.
The Ru–N2 (2.105(2) Å) bond length in complex 2 is longer than the Ru–N2 bond length in fac- and mer,trans-[RuCl3(NO)(PMA)] (2.2155(18) and 2.222(3) Å, respectively [7] ), in agreement with the presence of a Ru–Nsp2 bond in complex 2 as compared with the Ru–Nsp3 bond in fac- and mer,trans-[RuCl3(NO)(PMA)].
The Ru–NO (1.750(2) Å) and the N–O (1.135(3) Å) bond lengths are in the range found for these same bond lengths in other fac- and mer-[RuCl3(NO)(L–L)] (L–L = P–N or P–P donor ligands) complexes [7, 9, 40, 41]. Similarly, the average Ru–Cl bond length agrees with the Ru–Cl bond lengths described for other complexes in the literature [7, 9, 28].
The Ru–N–O bond angle in complex 2 is 168.4(3)°, whereas this same bond angle is 163.7(3)° in mer,trans-[RuCl3(NO)(PMA)] [7]. This value is compatible with the fact that PHOX is a less basic ligand than PMA. A more basic ligand should increase the Ru3+–NO0 character and lead to smaller νNO for mer,trans-[RuCl3(NO)(PMA)] as compared with complex 2. In addition, this difference agrees with the longer N–O bond length in mer,trans-[RuCl3(NO)(PMA)] (1.148(4) Å) as compared with mer,trans-[RuCl3(NO)(PHOX)] (1.133(4) Å).
Cyclic voltammetry
Cyclic voltammetry studies on complexes 1 and 2 (see Fig. 3 for the cyclic voltammograms of complex 1, which shows the processes Ru2+–NO+/Ru2+–NO0 and Ru2+–NO+/Ru3+–NO+), revealed one monoelectronic and irreversible redox process attributed to RuII–NO+ → RuII–NO0. The process occurred at −0.71 and −0.62 V in dichloromethane for complexes 1 and 2, respectively. Cyclic voltammetry conducted in acetonitrile showed that the reduction potentials shifted to −0.41 and −0.44 V for complexes 1 and 2, respectively. The electrochemical behavior of complexes 1 and 2 as well as the potential values resembled the values observed for other complexes containing “RuCl3(NO)” [7, 9, 28, 35].

Cyclic voltammograms of fac-[RuCl3(NO)(PHOX)] (1), in CH3CN (PTBA 0.1 M, scan rate = 100 mV s−1), showing a Ru2+–NO+/Ru2+–NO0 process and b Ru2+–NO+/Ru3+–NO+ process
The LUMO orbitals are mostly centered on NO+ [7, 34] and are the antibonding molecular orbitals of the Ru → NO π-bond overlap. In this way, the reduction process will be mainly centered on NO+, and the potential values will be largely influenced by the solvent, as described above and highlighted in Table 2. The difference between the reduction potentials of the fac and mer complexes was higher in dichloromethane than in acetonitrile.
In dichloromethane, the reduction potential of complex 1 (fac isomer) was more negative than the reduction potential of complex 2 (mer isomer). In acetonitrile, the reduction potential of both the fac and mer,trans isomers decreased and became similar, as observed for other similar complexes (see Table 2). Acetonitrile probably exerted some leveling effect on the reduction potentials during the cyclic voltammetry experiments. Considering νNO for the fac and mer complexes, complex 2 should undergo reduction at more negative potentials, because its nitrosyl should be less positive as compared with the nitrosyl in complex 1. On the other hand, the presence of a π-acceptor ligand (P atom of the PHOX) trans to the nitrosyl group should weaken the Ru → NO back bonding and lower the energy of the LUMO orbitals, facilitating the reduction process in complex 2 more than in complex 1.
The reduction potential value obtained for complex 1 was higher than the reduction potential values reported for the closest analogue fac-[RuCl3(NO)(PMA)] [7]. This can be rationalized in terms of the basicity of the N-donor of PMA as compared with the basicity of the N-donor of PHOX (see Table 2). The more basic ligand should increase the Ru → NO back bonding and raise the energy of the LUMO orbitals, making the reduction process more difficult (more negative potential values). The reduction potential value for complex 2 was the same of the reported for complex mer,trans-[RuCl3(NO)(PMA)] [7].
The ability of NO+ to withdraw electron density from ruthenium through Ru → NO back bonding shifts the potentials of the RuII → RuIII oxidation process to higher values in ruthenium nitrosyl complexes, even when compared with carbonyl-containing complexes [42]. For complexes 1 and 2, the RuII → RuIII oxidation process occurred at 2.27 and 2.04 V, respectively. The higher value observed for complex 1 as compared with complex 2 was due to the presence of a P atom trans to NO+, in complex 2, which increased the electron density over the ruthenium center and elevated the energy of the HOMO orbitals. These potential values were higher than the potential values described for the fac- and mer,trans-[RuCl3(NO)(PMA)] analogues. Two effects could explain these differences: (1) the σ-donor/π-acceptor properties of the N-donor group and (2) the size of the ring of the chelating agent. The PMA ligand has a strong σ-donor character (-N(CH3)2) as compared with the less basic PHOX ligand, so PMA should increase the electron density over ruthenium. On the other hand, the N-donor of the PHOX ligand should be able to act as a π-acceptor group, to decrease the electron density over ruthenium through back-bond interaction. Additionally, a five- and a six-membered ring should arise when the PMA and the PHOX ligands chelate to the metal center, respectively. A five-membered ring should stabilize the Ru3+ formed upon oxidation better than a six-membered ring.
Photo- and thermal isomerization 1 ↔ 2
As described earlier in this document, complex 1 underwent photochemical isomerization in dichloromethane, to form complex 2 in quantitative yield (determined by 31P{1H} and 1H NMR) after 3 days. We had already observed the same effect for fac-[RuCl3(NO)(PMA)], and we used 15N-labeled NO to obtain evidence of the isomerization mechanism [7]. On that occasion, we concluded that fac → mer isomerization did not occur via a dissociative pathway involving the Ru–NO bond cleavage.
The following mechanisms could explain the photo-isomerization of complex 1 into complex 2 in dichloromethane solution: a non-dissociative process via metastable linkage NO isomers (η1–O or η2–NO) [43] and two dissociative processes involving either Ru–Cl [44] or Ru–N(PHOX) bond dissociation/association [45]. Dissociation of the chloride would result in ionic species in solution, which would not be favorable in dichloromethane, due to the low relative polarity (0.309) and dielectric constant (9.08) of this solvent [46, 47]. Hence, the most plausible mechanisms for the photochemical process in dichloromethane would be the non-dissociative metastable linkage isomers or the dissociative process involving the Ru–N(PHOX) bond.
It is noteworthy that reflux of a suspension of complex 2 in methanol, but not in dichloromethane, gave complex 1 after 3 h. This observation was compatible with a dissociative mechanism involving Ru–Cl bond cleavage; methanol is a polar protic solvent (relative polarity = 0.762 and dielectric constant = 32.6 [46, 47]) and could stabilize the resulting ionic species. These ionic species would hardly arise in dichloromethane, and thermal isomerization should not occur in this solvent.
To verify the possible dissociation of the chloride, we conducted thermal isomerization of complex 2 in the presence of KCl at a complex 2/KCl molar ratio of 1:5. After refluxing the methanol suspension for 3 h, no visual change took place (it is important to bear in mind that complex 2 is green and complex 1 is orange). 1H and 31P{1H} NMR spectra recorded after evaporation of the solvent to dryness under reduced pressure followed by extraction of the solid residue with deuterated dichloromethane CD2Cl2 and purification on a Celite column revealed that thermal isomerization was suppressed and less than 5 % of complex 1 emerged. These results corroborated the major contribution of the dissociative mechanism involving the Ru–Cl bond dissociation/association.
Conclusion
This work has reported the successful synthesis and characterization of complexes fac- and mer,trans-[RuCl3(NO)(PHOX)] (complexes 1 and 2, respectively). Single-crystal X-ray diffractometry helped to solve the molecular structure of complex 2. The chemical behavior of these complexes resembled the behavior of other P–N and P–P analogues published by us and other research groups. Complex 1 isomerized to complex 2 in dichloromethane under ambient light. Reflux of a methanol suspension of complex 2 reversed the process. A plausible mechanism for the photo-isomerization of complex 1 to complex 2 is a dissociative pathway involving Ru–N(PHOX) bond dissociation/association. The thermal isomerization of complex 2 to complex 1 indicated that the dissociation/association of one Ru–Cl bond dominates the mechanism, because addition of KCl inhibits the process, which in turn does not occur in dichloromethane. We are currently investigating the mechanism of the photo- and thermal isomerization of complexes with general formula [RuCl3(NO)(L–L)] (L–L = P–N or P–P), including DFT calculations on the intermediates.
Supplementary material
Crystallographic data (excluding structure factors) for complex 2 have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication on CCDC 1038574. Copies of the data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033; or e-mail: deposit@ccdc.cam.ac.uk).
References
Braunstein P (2004) J Organomet Chem 689(24):3953–3967
Naud PBAF (2001) Angewandte Chemie 40:680–699
Rauchfuss TB, Patino FT, Roundhill DM (1975) Inorg Chem 14(3):652–656
Mudalige DC, Ma ESF, Rettig SJ, James BR, Cullen WR (1997) Inorg Chem 36(24):5426
Ma ESF, Rettig SJ, James BR (1999) Chemical Communications 24:2463–2464
Pamplin CB, Ma ESF, Safari N, Rettig SJ, James BR (2001) J Am Chem Soc 123(35):8596–8597
da Silva JP, Caetano FR, Cavarzan DA, Fagundes FD, Romualdo LL, Ellena J, Jaworska M, Lodowski P, Barison A, de Araujo MP (2011) Inorg Chim Acta 373(1):8–18
Fagundes FD, da Silva JP, Veber CL, Barison A, Pinheiro CB, Back DF, de Sousa JR, de Araujo MP (2012) Polyhedron 42(1):207–215. doi:10.1016/j.poly.2012.05.016
Cavarzan DA, Caetano FR, Romualdo LL, do Nascimento FB, Batista AA, Ellena J, Barison A, de Araujo MP (2006) Inorg Chem Commun 9(12):1247–1250
Kunz PC, Thiel I, Noffke AL, Reiß GJ, Mohr F, Spingler B (2012) J Organomet Chem 697(1):33–40
Aliende C, Pérez-Manrique M, Jalón FA, Manzano BR, Rodríguez AM, Cuevas JV, Espino G, Martínez MÁ, Massaguer A, González-Bártulos M, de Llorens R, Moreno V (2012) J Inorg Biochem 117:171–188
García-Álvarez R, García-Garrido SE, Díez J, Crochet P, Cadierno V (2012) Eur J Inorg Chem 2012(26):4218–4230
Wajda-Hermanowicz K, Ciunik Z, Kochel A (2006) Inorg Chem 45(8):3369–3377
Curvey N, Widaman AK, Rath NP, Bauer EB (2014) Tetrahedron Lett 55(19):3033–3037
Albrecht C, Gauthier S, Wolf J, Scopelliti R, Severin K (2009) Eur J Inorg Chem 2009(8):1003–1010
Langer T, Helmchen G (1996) Tetrahedron Lett 37(9):1381–1384
Wang YL, Liu DL, Meng QH, Zhang WB (2009) Tetrahedron-Asymmetry 20(21):2510–2512
Fritz HP, Gordon IR, Schwarzhans KE, Venanzi LM (1965) 969. J Chem Soc (Resumed):5210–5216
Mann FG, Watson J (1948) J Org Chem 13(4):502–531
Tolmachev AA, Yurchenko AA, Merculov AS, Semenova MG, Zarudnitskii EV, Ivanov VV, Pinchuk AM (1999) Heteroat Chem 10(7):585–597
Peer M, de Jong JC, Kiefer M, Langer T, Rieck H, Schell H, Sennhenn P, Sprinz J, Steinhagen H, Wiese B, Helmchen G (1996) Tetrahedron 52(21):7547–7583
da Silva JP, Fagundes FD, Back DF, Ellena J, de Araujo MP (2015) Cationic ruthenium-nitrosyl complexes with general formula [RuCl2(NO)(P-N)(PR3)]+: effect of PR3 ligands. Polyhedron (submitted)
Fergusson JE, Coll RK (1993) Inorg Chim Acta 207(2):191–197
Innorta G, Foffani A, Torroni S, Serrazanetti G (1979) Inorg Chim Acta 35:189–196
Chatt J, Shaw BL (1966) J Chem Soc A Inorg Phys Theor 0:1811–1812
Correa RS, Barolli JP, Barbosa MIF, Ellena J, Batista AA (2013) J Mol Struct 1048:11–17
Von Poelhsitz G, Bogado AL, de Araujo MP, Selistre-de-Araujo HS, Ellena J, Castellano EE, Batista AA (2007) Polyhedron 26(16):4707–4712
Von Poelhsitz G, de Lima RC, Carlos RM, Ferreira AG, Batista AA, de Araujo AS, Ellena J, Castellano EE (2006) Inorg Chim Acta 359(9):2896–2909
Sheldrick GM (2008) Acta Cryst. A64:112–122
Fletcher JM, Jenkins IL, Lever FM, Martin FS, Powell AR, Todd R (1955) J Inorg Nucl Chem 1(6):378–401
Wüstenberg B, Pfaltz A (2008) Adv Synth Catal 350(1):174–178
Coe BJ, Glenwright SJ (2000) Coord Chem Rev 203:5–80
Queiroz SL, Batista AA, Oliva G, Gambardella M, Santos RHA, MacFarlane KS, Rettig SJ, James BR (1998) Inorg Chim Acta 267(2):209–221
Lopes LGF, Castellano EE, Ferreira AG, Davanzo CU, Clarke MJ, Franco DW (2005) Inorg Chim Acta 358(10):2883–2890
Von Poelhsitz G, de Araujo MP, de Oliveira LAA, Queiroz SL, Ellena J, Castellano EE, Ferreira AG, Batista AA (2002) Polyhedron 21(22):2221–2225
Garrou PE (1981) Chem Rev 81(3):229–266
Garrou PE (1975) Inorg Chem 14(6):1435–1439
Aullon G, Alvarez S, Cao R, Ortiz M, Diaz-Garcia AM (2009) Inorg Chim Acta 362(12):4651–4658
Calandreli I, Oliveira FD, Liang GG, da Rocha ZN, Tfouni E (2009) Inorg Chem Commun 12(7):591–595
Golfeto CC, Von Poelhsitz G, Selistre-de-Araujo HS, de Araujo MP, Ellena J, Castellano EE, Lopes LGL, Moreira IS, Batista AA (2010) J Inorg Biochem 104(5):489–495
Von Poelhsitz G, Bogado AL, Liao LM, Ferreira AG, Castellano EE, Ellena J, Batista AA (2010) Polyhedron 29(1):280–287
Cavarzan DA, Fagundes FD, Fuganti O, da Silva CWP, Pinheiro CB, Back DF, Barison A, Bogado AL, de Araujo MP (2013) Polyhedron 62:75–82
Coppens P, Novozhilova I, Kovalevsky A (2002) Chem Rev 102(4):861–883
Nguyen DH, Daran J-C, Mallet-Ladeira S, Davin T, Maron L, Urrutigoity M, Kalck P, Gouygou M (2013) Dalton Trans 42(1):75–81
Gavriluta A, Büchel GE, Freitag L, Novitchi G, Tommasino JB, Jeanneau E, Kuhn P-S, González L, Arion VB, Luneau D (2013) Inorg Chem 52(11):6260–6272
Reichardt C (2004) Empirical parameters of solvent polarity. In: Reichardt C (ed) Solvents and solvent effects in organic chemistry. Wiley-VCH Verlag GmbH & Co. KGaA, London, pp 389–469
Lide DR (ed) (2006) Handbook of chemistry and physics, 87th edn. CRC, Boca Raton
Acknowledgments
The authors are grateful to CNPq, CAPES (PROCAD/NF 2009 and CAPES/UdelaR 2010), FINEP, and NENNAM (PRONEX F. Araucária/CNPq protocolo 17378) for financial support, and to Johnson Matthey plc for the loan of RuCl3 (to M.P.A).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
da Silva, J.P., Lordello, L.D., de Oliveira, A.R.M. et al. Ruthenium nitrosyl complexes [RuCl3(NO)(P–N)] bearing an oxazoline-derived P–N ligand (PHOX). Transition Met Chem 40, 363–370 (2015). https://doi.org/10.1007/s11243-015-9925-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-015-9925-2