
Abstract
The copper aminotropones Cu[ON(R′)C7H4R-4]2 [R = H, R′ = Me (13), Et (14), n-Pr (15), n-Bu (16), Bz (17), MenOCH2CH2 (20); R = i-Pr, R′ = Me (18), n-Pr (19), MenOCH2CH2 (21)] have been prepared from the corresponding aminotropones HN(R′)OC7H4R-4 (1–7) by reacting with copper(II) acetate in aqueous ethanol. 20, 21 contain the flavourant, menthol, as part of the ligand. The structures of 5 (R = H, R′ = Bz), a hydrogen-bonded dimer, 14 and 20, both incorporating square-planar, four-coordinate copper centres, have been determined by X-ray crystallography. The antibacterial activities of complexes 13, 17, 20 and 21 have been assayed against Staphylococcus waneri, an in vitro model of plaque inhibition effects, and found to be more active than a commercial toothpaste formulation, but less active than the O,O-chelated copper(II) complex of ethylmaltol.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Hinokitiol, a naturally occurring α-hydroxyketone which can be extracted from wood (Thujapalicata, Taiwan Hinoki) continues to attract interest as a ligand due to its attractive biological properties (antimicrobial [1, 2], insecticidal [3], cytotoxicity on tumour cells [4]) which make it a favourable chelating agent for metals which might offer biological synergism. As a result, numerous metal derivatives of both hinokitiol (I, R = i-Pr) [5], and its parent, tropolone (I, R = H) [6–11], have been prepared and analysed. More recently, tropolone has been elaborated to form mesogenic complexes when coordinated to metals [12, 13]. Our own work in this area stems from an interest in copper, zinc and tin complexes which can be incorporated into dental formulations [14], including complexes of this triad with ligands such as maltol and ethylmaltol as well as I [15–17]. In this regard, the ligand must be orally acceptable, be able to deliver the metal in the correct oxidation state and concentration (i.e. soluble complexes) and have reasonable long-term stability.
As part of this general interest, we have become interested in the related 2-(alkylamino)tropone ligand system (II), which offers a complimentary κ2-O,N chelation mode to the κ2-O,O mode inherent in I. Relatively few reports have appeared concerning the chelation of metals such as Cu(II) [18–20] and Zn(II) [21] with II, to which we now add our own findings relating to copper. In particular, as well as broadening the scope of simple ML2 complexes, we report on the incorporation of a flavouring component, menthol, onto the ligand periphery. As one of the drawbacks of these complexes for oral delivery is their metallic taste even at low concentrations, this approach may go some way to mitigating this problem. 
Experimental
Starting materials: tropolone was obtained commercially from Avocado, and hinokitiol was provided by Unilever Research; both were used as received without further purification. Reactions were carried out in air unless otherwise specified. Elemental analyses were performed on a Carlo-Erba Strumentazione E. A. model 1106 microanalyser, and the temperature of the furnace was set to 500 °C. The results were duplicated, and the mean of the duplicated measurements was the final result. All 1H and 13C NMR spectra were recorded on a Bruker Avance (300 MHz) Fourier transform spectrometer. All spectra were recorded in d-chloroform and are referenced to residual solvent peaks. Peak positions were recorded in δ ppm with abbreviations s, d, t and m denoting singlet, doublet, triplet and multiplet, respectively. All coupling constants (J) are quoted in Hertz. Infrared spectra were recorded as a thin oil film sandwiched between NaCl plates in the frequency range 4,000–400 cm−1 on a Nicolet 510P FT-IR spectrometer. All absorptions are quoted in cm−1.
Synthesis of 2-p-toluenesulfonyltropolone
Following a previously reported procedure [22], tropolone (5.00 g, 41.0 mmol) and p-toluenesulfonyl chloride (7.81 g, 41.0 mmol) were mixed together in dry pyridine (20 mL). After 30 min, the mixture was filtered and the product obtained was then washed with cold water and dried in vacuo, yielding the product as a white solid (11.18 g, 98 %). Analysis: Found [calc. for C14H12O4S]: C 61.0 (60.9) %; H 4.4 (4.3) %. M. p.: 156–158 °C (lit: 156.5–157.5 °C) [23].
Synthesis of 2-p-toluenesulfonylhinokitiol
The method described above [22] was applied to hinokitiol (5.00 g, 30.5 mmol). Hinokitiol and p-toluenesulfonyl chloride (5.81 g, 30.5 mmol) were mixed in dry pyridine (20 mL); the product was then washed with cold water and dried in vacuo, to obtain a pale yellow solid (9.31 g, 96 %). Analysis: Found (calc. for C17H18O4S): C 64.6 (64.2) %; H 6.1 (5.7) %.
The 2-alkylaminotropones and 2-alkylaminohinokitiols were prepared following literature procedures; [23, 24] the methodology described by Rasika Dias et al. [23] was used for (1), (2), (6), (7), and the one from Pietra et al. [24] was repeated for (3)–(5) and (8)–(10).
Synthesis of 2-methylaminotropone (1)
2-Methylaminotropone was prepared using a solution of 2-p-toluenesulfonyltropolone (2.00 g, 7.24 mmol) in dichloromethane (30 mL) and aqueous methylamine (40 %) (10 mL). The mixture was stirred overnight at room temperature, then the two layers were separated, and the organic layer was washed with water, dried with magnesium sulphate, and concentrated under vacuum to obtain a yellow solid (0.76 g, 78 %). Analysis: Found (calc. for C8H9NO): C 61.0 (60.9); H 4.4 (4.3); N 4.1 (4.0) %. 1H NMR [δ(ppm), CDCl3]: 2.95 [3H, s, NCH 3]; 6.50–7.19 [5H, m, C7 H 5]. 13C NMR [δ(ppm), CDCl3]: 28.5 [CH3]; 107.3, 121.0, 127.3, 135.3, 136.3 [CH];155.5 [C 2]; 175.6 [C 3]. IR: 3,293 υ(N–H); 1,629, 1,590 υ(C=O); 1,510 υ(C=C).
2-Ethylaminotropone (2)
The methodology described above was employed with aqueous ethylamine (70 %) (10 mL) and 2-p-toluenesulfonyltropolone (2.00 g, 7.24 mmol) in dichloromethane (30 mL). An orange oil was obtained (0.77 g, 71 %). Analysis: Found (calc. for C9H11NO): C 71.9 (72.5); H 7.4 (7.4); N 9.3 (9.4) %. 1H NMR [δ(ppm), CDCl3]: 1.31 [3H, t, NCH2CH 3]; 3.28 [2H, q, NCH2CH 3]; 6.49–7.21 [5H, m, C7 H 5 ]. 13C NMR [δ(ppm), CDCl3]: 13.4 [CH3]; 37.2 [CH2]; 108.4, 121.7, 128.0, 136.1, 136.9 [CH]; 155.2 [C 2]; 176.2 [C 3]. IR data g,: 3,301 υ(N–H); 1,630, 1,594 υ(C=O); 1,515 υ(C=C).
2-n-Propylaminotropone (3)
To a solution of 2-p-toluenesulfonyltropolone (2.00 g, 7.24 mmol) in dried DMSO (100 mL) was added n-propylamine (10 mL), and the solution was stirred overnight. The mixture was poured into water and extracted with ether; the ether layer was dried with magnesium sulphate; and the solvent removed in vacuo. A brown oil was obtained (0.81 g, 68 %). Analysis: Found [calc. for C10H13NO]: C 72.7 (73.6); H 8.0 (8.0); N 8.65 (8.58) %. 1H NMR [δ(ppm), CDCl3]: 0.94 [3H, t, NCH2CH2CH 3]; 1.64 [2H, m, NCH2CH 2CH3]; 3.18 [2H, m, NCH 2CH2CH3]; 6.49–7.21 [5H, m, C7 H 5]. 13C NMR [δ(ppm), CDCl3]: 12.1 [CH2CH2 CH3]; 22.2 [CH2 CH2CH3]; 44.9 [CH2CH2CH3]; 108.8, 122.1, 128.4, 136.5, 137.3 [CH]; 155.7 [C 2]; 176.5 [C 3]. IR: 3,291 υ(N–H); 1,637, 1,595 υ(C=O); 1,512 υ(C=C).
2-n-Butylaminotropone (4)
Following the procedure with n-butylamine (10 mL) and 2-p-toluenesulfonyltropolone (2.00 g, 7.24 mmol), 4 was obtained (0.89 g, 69 %) as a brown-orange oil. Analysis: Found [calc. for C11H15NO]: C 72.6 (74.6); H 8.6 (8.5); N 7.9 (7.9) %. 1H NMR [δ(ppm), CDCl3]: 0.94 [3H, t, NCH2CH2CH2CH 3]; 1.40 [2H, m, NCH2CH2CH 2CH3]; 1.63 [2H, m, NCH2CH 2CH2CH3]; 3.22 [2H, t, NCH 2CH2CH2CH3]; 6.49–7.21 [5H, m, C7 H 5 ]. 13C NMR [δ(ppm), CDCl3]: 13.5 [CH2CH2CH2 CH3]; 20.0 [CH2CH2 CH2CH3]; 30.2 [CH2 CH2CH2CH3]; 42.3 [CH2CH2CH2CH3]; 108.4, 121.7, 127.9, 136.1, 136.9 [CH]; 155.4 [C 2]; 176.2 [C 3]. IR: 3,307 υ(N–H); 1,636, 1,593 υ(C=O); 1,516 υ(C=C).
2-Benzylaminotropone (5)
Following the same method as for (3), a yellow solid was obtained using benzylamine (10 mL) and 2-p-toluenesulfonyltropolone (2.00 g, 7.24 mmol). Yield 1.34 g, 88 %. Recrystallisation from chloroform/cyclohexane gave yellow crystals suitable for X-ray analysis. Analysis: Found [calc. for C14H13NO]: C 79.5 (79.6); H 6.3 (6.2); N 6.7 (6.6) %. 1H NMR [δ(ppm), CDCl3]: 4.44 [2H, s, CH 2]; 6.43–7.52 [10H, m, C6 H 5 and C7 H 5]. 13C NMR [δ(ppm), CDCl3]: 46.5 [CH2]; 108.4, 122.2, 127.5, 128.0, 130.1, 131.4, 136.1, 136.9 [CH]; 156.0 [C 2]; 175.8 [C 3]. IR: 3,304 υ(N–H); 1,632, 1,592 υ(C=O); 1,512 υ(C=C).
2-Methylamino-4-i-propyltropone (6)
The method described earlier [23] for (1) was repeated for (6) using 2-p-toluenesulfonylhinokitiol (2.00 g, 6.28 mmol) and the corresponding amine in excess in solution in dichloromethane (30 mL). Aqueous methylamine (40 %) (10 mL) was used for (6) (0.57 g, 51 %), aqueous ethylamine (70 %) (10 mL) for (7) (0.92 g, 76 %). Analysis for (6): Found [calc. for C11H15NO]: C 72.4 (74.6); H 8.3 (8.5); N 7.6 (7.9) %. IR: 3,286 υ(N–H); 1,633, 1,595 υ(C=O); 1,514 υ(C=C).
2-n-Propylamino-4-i-propyltropone (7)
Following the procedure for (3), (7) was obtained using 2-p-toluenesulfonylhinokitiol (2.00 g, 6.28 mmol) and npropylamine (10 mL). Yields 0.51 g, 39 %. Analysis for (8): Found [calc. for C13H19NO]: C 74.0 (76.1); H 9.5 (9.3); N 6.2 (6.8). IR: 3,303 υ(N–H); 1,632, 1,592 υ(C=O); 1,511 υ(C=C).
Synthesis of menthoxyacetic acid (8)
Following a previously reported procedure [25], menthol (15.6 g, 100 mmol) was dissolved in THF (ca. 50 mL) under nitrogen. To this solution was added lithium ribbon (0.80 g, 115 mmol) cut into small pieces, and this mixture was refluxed under nitrogen for 4 h. This solution was then filtered to remove the unreacted lithium, and dry monochloroacetic acid (4.25 g, 45.1 mmol) in anhydrous THF (12.5 mL) was added dropwise to the filtrate. This mixture was gently refluxed for 20 h. After this time, water (ca. 30 mL) was added. The THF and menthol were fractionally distilled from the yellow suspension, which was then cooled in an ice bath and filtered to obtain the solid lithium salt as a white precipitate. This was dissolved in dilute HCl and extracted using diethyl ether. The resulting ethereal solution was washed with water, dried over magnesium sulphate, filtered, and concentrated under rotary evaporation to produce a yellow oil (2.25 g; 10 %). 1H NMR (CDCl3): 8.10 [H, br s, OH]; 4.13 [1H, d, Ja-b = 15.0 Hz, C2 H a]; 4.02 [1H, d, Jb-a = 15.0 Hz, C2 H b]; 3.13 [1H, ddd, J3-4ax = J3-8 = 9.10, J3-4eq = 3.00 Hz, C3 H]; 1.10–2.30 [2H, m, C4 H 2 ; 1H, m, C5 H; 2H, m, C6 H 2 ; 2H, m, C7 H 2 ; 1H, m, C8 H; 1H, m, C9 H]; 0.83, 0.75 [3H, d, J10-9/J11-9 = 6.30, 8.40 Hz, C10 H 3 /C11 H 3 ]; 0.85 [3H, d, J12-5 = 8.10 Hz, C12 H 3 ]. 13C NMR (CDCl3): 176 [C 1]; 66.3 [C 2]; 80.8 [C 3]; 40.2 [C 4]; 31.9 [C 5]; 34.7 [C 6]; 22.6 [C 7]; 48.3 [C 8]; 26.0 [C 9]; 21.4, 16.4 [C 10 /C 11]; 23.5 [C 12]. IR: 1,720ν (C=O); 1,275 ν(C–O); 3,120 ν(O–H).
Synthesis of 2-menthoxyacetamide (9) [26]
Menthxoyacetyl chloride was produced by dissolving menthoxyacetic acid (8) (10.0 g, 46.7 mmol) in thionyl chloride (16.7 mL, 229 mmol) and warming at 50 °C for 3 h. The excess thionyl chloride was removed by warming in a water bath under reduced pressure, and the acid chloride (7.30 g; 67 %) was used directly. Ammonia (19.2 mL) was added directly to the acid chloride, (7.30 g, 31.3 mmol) and stirred at 0 °C for 24 h. The final product was washed with water, extracted with diethyl ether (2 × 20 mL), dried over anhydrous magnesium sulphate, filtered, and dried in vacuo yielding a white solid. Recrystallisation from petroleum-ether 60–80 °C gave white crystals (4.58 g; 46 %; m. p. (°C): 92–94 (Lit: 92–94 [26]). Analysis for (22): Found [calc. for C12H23O2N]: C 66.4 (67.7); H 10.7 (10.7); N 6.2 (6.5) %. 1H NMR (CDCl3): 5.80, 6.59 [2H, br s, NH 2 ]; 3.98 [1H, d, Ja-b = 15.0 Hz, C2 H a; 3.78 [1H, d, Jb-a = 15.6 Hz, C2 H a]; 3.10 [1H, ddd, J3-4ax = J3-8 = 11.4, J3-4eq = 4.50 Hz, C3 H]; 1.20–2.20 [2H, m, C4 H 2 ; 1H, m C5 H; 2H, m, C6 H 2 ; 2H, m, C7 H 2 ; 1H, m, C8 H; 1H, m, C9 H]; 0.84, 0.72 [3H, d, J10-9/J11-9 = 3.90, 6.60 Hz, C10 H 3 /C11 H 3 ]; 0.87 [3H, d, J12-5 = 3.30 Hz, C12 H 3 ]. 13C NMR (CDCl3): 174 [C 1]; 68.1 [C 2]; 80.8 [C 3]; 40.5 [C 4]; 31.8 [C 5]; 34.8 [C 6]; 22.6 [C 7]; 48.4 [C 8]; 26.4 [C 9]; 21.3, 16.6 [C 10/C 11]; 23.6 [C 12]. IR: 3,189 ν(N–H); 1,640 ν(C=O).
Synthesis of 2-menthoxyethylamine (10) [26]
In a three-necked round-bottomed flask were placed 2-menthoxyacetamide (9) (4.01 g, 18.9 mmol) and anhydrous THF (30 mL). Lithium aluminium hydride (2.15 g, 56.7 mmol) dissolved in anhydrous THF (20 mL) was added dropwise using a syringe under the flow of nitrogen to the 2-menthoxyacetamide. During the addition, the reaction mixture gave off bubbles and became grey. The contents of the flask were stirred vigorously and allowed to reflux for 8 h, then cooled to room temperature, and stirred at this temperature overnight. The mixture was cooled to 0 °C by application of an ice bath, and water (5 mL) was added dropwise. A white precipitate was produced, and bubbles of gas were given off. The precipitate was removed by filtration and washed with diethyl ether. The filtrate was extracted with diethyl ether (2 × 25 mL) and 15 % (w/v) sodium hydroxide (5 mL). The organic layers were combined, dried over anhydrous magnesium sulphate, filtered, and concentrated to dryness by rotary evaporation yielding a colourless liquid (2.87 g; 78 %). Analysis for (23): Found (calc. for C12H25ON): C 70.1 (72.4); H 12.2 (12.7); N 5.2 (5.4) %. 1H NMR (CDCl3): 2.75 [2H, t, JNH-1 = 3.00 Hz, NH 2 ]; 3.65 [2H, m, C1 H 2 ]; 3.25 [2H, m, C2 H 2 ]; 2.97 [1H, ddd, J3-4ax = J3-8 = 10.5, J3-4eq = 4.50 Hz, C3 H]; 1.15–2.20 [2H, m, C4 H 2 ; 1H, m, C5 H; 2H, C6 H 2 ; 2H, m, C7 H 2 ; 1H, m, C8 H; 1H, m, C9 H]; 0.83, 0.71 [3H, d, J10-9/J11-9 = 6.60, 7.20 Hz, C10 H 3 /C11 H 3 ]; 0.86 [3H, d, J12-5 = 6.00 Hz, C12 H 3 ]. 13C NMR (CDCl3): 39.5 [C 1]; 69.6 [C 2]; 78.3 [C 3]; 41.3 [C 4]; 30.5 [C 5]; 33.6 [C 6]; 21.4 [C 7]; 47.3 [C 8]; 26.7 [C 9]; 19.9, 15.2 [C 10 /C 11]; 22.3 [C 12]. IR: 3,205 ν(N–H).
Synthesis of 2-menthoxyethylamino-cyclohept-2,4,6-trien-1-one.H2O (11)
2-Menthoxyethylamino-cyclohept-2,4,6-trien-1-one.H2O (11) was prepared by reacting 2-menthoxyethylamine (10) (1.00 g, 5.03 mmol), two equivalents of triethylamine (0.93 mL, 6.67 mmol) and 2-p-toluenesulfonyltropolone (0.92 g, 3.33 mmol) in DMSO (50 mL). This mixture was stirred for 1 week at room temperature. After completion of the reaction, the reaction mixture was dissolved in water and the two layers were separated using diethyl ether. The organic layer was washed with water, dried over magnesium sulphate, filtered, and rotary evaporated to obtain a brown oil. Concentrated HCl was added dropwise to this mixture until the pH reached 1. This was extracted with diethyl ether and washed with water (2 × 50 mL), dried over magnesium sulphate, filtered, and rotary evaporated to yield a brown oil (0.96 g; 60 %). Analysis for (11): Found [calc. C19H31O3N]: C 71.2 (71.0); H 9.9 (9.7); N 4.4 (4.4) %. 1H NMR (CDCl3): 7.20–6.38 [5H, m, C3 H, C4 H, C5 H, C6 H, C7 H]; 3.55 [1H, t, NH]; 3.32–3.90 [4H, m, C8 H 2 , C9 H 2 ]; 3.01 [1H. ddd, J10-11ax = J10-15 = 10.5, J10-11eq = 4.50 Hz, C10 H]; 1.15-2.20 [2H, m, C11 H 2 ; 1H, m, C12 H; 2H, m, C13 H 2 ; 2H, m, C14 H 2 ; 1H, m, C15 H; 1H, m, C16 H]; 0.82, 0.70 [3H, d, J17-16/J18-16 = 6.60, 7.50 Hz, C17 H 3 /C18 H 3 ]; 0.85 [3H, d, J19-12 = 6.30 Hz, C19 H 3 ]. 13C NMR (CDCl3): 177 [C 1]; 156 [C 2]; 123 [C 3]; 137 [C 4]; 129 [C 5]; 137 [C 6]; 108 [C 7]; 66.3 [C 8]; 40.8 [C 9]; 80.3 [C 10]; 43.5 [C 11]; 31.9 [C 12]; 34.8 [C 13]; 21.8 [C 14]; 48.6 [C 15]; 26.1 [C 16]; 21.3, 16.6 [C 17/C 18]; 23.7 [C 19].IR: 3,300 ν(N–H) and ν(O–H); 1,631, 1,595 ν(C=O); 1,519 ν(C=C).
Synthesis of 2-menthoxyethylamino-4-isopropylcyclohept-2,4,6-trien-1 one.H2O (12)
2-Menthoxyethylamino-4-isopropylcyclohept-2,4,6-trien-1-one.H2O (12) was prepared in a similar manner to 11 by reacting 2-menthoxyethylamine (10) (2.00 g, 10.1 mmol), two equivalents of triethylamine (1.86 mL, 13.3 mmol) and 2-p-toluenesulfonylhinokitiol (2.12 g, 6.67 mmol) in DMSO (50 mL) to yield the product as a brown oil (2.54 g; 70 %). Analysis for (12): Found [calc. C22H37O3N]: C 73.1 (72.7); H 10.1 (10.2); N 3.2 (3.8) %. 1H NMR (CDCl3): 7.45–6.28 [4H, m, C3 H, C5 H, C6 H, C7 H]; 3.45 [1H, t, NH]; 3.30–3.95 [4H, m, C8 H 2 , C9 H 2 ]; 3.01 [1H. ddd, J10-11ax = J10-15 = 10.5, J10-11eq = 4.50 Hz, C10 H]; 1.20–2.20 20 [2H, m, C11 H 2 ; 1H, m, C12 H; 2H, m, C13 H 2 ; 2H, m, C14 H 2 ; 1H, m, C15 H; 1H, m, C16 H]; 0.83, 0.70 [3H, d, J17-16/J18-16 = 6.00, 7.20 Hz, C17 H 3 /C18 H 3 ]; 0.81 [3H, d, = 9.00 Hz,]; 0.83 [3H, d, J19-12 = 5.90 Hz, C19 H 3 ]; 2.80 [1H, tt, C20 H]; 1.15 [3H, d, C21 H 3 /C22 H 3 ,]. 13C NMR (CDCl3): 174 [C 1]; 158 [C 2]; 122 [C 3]; 135 [C 4]; 127 [C 5]; 137 [C 6]; 107 [C 7]; 68.5 [C 8]; 37.8 [C 9]; 78.6 [C 10]; 39.5 [C 11]; 30.5 [C 12]; 33.5 [C13]; 22.6 [C14]; 47.2 [C15]; 24.7 [C16]; 19.9, 15.5 [C17/C18]; 22.9 [C19]; 39.8 [C20]; 24.6, 24.5 [C21/C22]. IR: 3,295 ν(N–H); 1,635, 1,593 ν(C=O); 1,513 ν(C=C).
Synthesis of copper compounds
The method described by us previously [17] was adapted to prepare (13)–(21). A solution of the appropriate ligand in water/ethanol mix (1:1) was added to a well-stirred solution of copper(II) acetate in water/ethanol mix (1:1). After stirring for 2 h, the solution was refluxed and stirred for another 2 h and then left at room temperature.
Cu(2-methylaminotroponate)2 (13)
A dark green solid was obtained using (1) (0.81 g, 5.99 mmol) and copper acetate (0.60 g, 2.99 mmol). Yield 99 % (0.98 g). Recrystallisation was from ethanol. Analysis for (13): Found [calc. for C16H16N2O2Cu]: C 57.9 (57.9); H 4.86 (4.82); N 8.40 (8.44) %. IR: 1,596, 1,569 υ(C=O); 1,518 υ(C=C).
Cu(2-ethylaminotroponate)2 (14)
Following the same procedure using (2) (0.40 g, 2.68 mmol) and copper acetate (0.27 g, 1.34 mmol), (14) was obtained. Recrystallisation from chloroform yielded green crystals (0.46 g, 95 %) suitable for X-ray analysis. Analysis for (14): Found [calc. for C18H20N2O2Cu]: C 60.1 (60.0); H 5.7 (5.6); N 7.7 (7.8) %. IR: 1,595, 1,540 υ(C=O); 1,513 υ(C=C).
Cu(2-n-propylaminotroponate)2 (15)
Using the above methodology with (3) (0.55 g, 2.56 mmol) added to copper acetate (0.26 g, 1.28 mmol). Following this, a green solid was obtained (0.55 g, 86 %). Analysis for (15): Found [calc. for C20H24N2O2Cu]: C 63.1 (63.5); H 4.8 (4.9); N 5.8 (5.7) %. IR: 1,597, 1,570 υ(C=O); 1,516 υ(C=C).
Cu(2-benzylaminotroponate)2 (16)
Using the above methodology with (4) (0.29 g, 1.37 mmol) and copper acetate (0.14 g, 0.68 mmol). A green solid was obtained (0.32 g, 95 %). Analysis for (16): Found [calc. for C28H24N2O2Cu]: C 68.9 (69.5); H 5.1 (5.0); N 5.8 (5.8) %. IR: 1,593, 1,570 υ(C=O); 1,513 υ(C=C).
Cu(2-n-butylaminotroponate)2 (17)
A green solid was obtained using (5) (0.46 g. 2.59 mmol) and copper acetate (0.26 g, 1.29 mmol). Yield 0.46 g, 86 %. Analysis for (17): Found [calc. for C22H28N2O2Cu]: C 63.2 (63.5); H 6.8 (6.7); N 6.8 (6.7) %. IR: 1,597, 1,569 υ(C=O); 1,515 υ(C=C).
Cu(2-methylamino-4-i-propyltroponate)2 (18)
A green solid was obtained (0.21 g, 42 %) from (6) (0.50 g, 2.82 mmol) added to copper acetate (0.28 g, 1.41 mmol). Analysis for (18): Found [calc. for C22H28N2O2Cu]: C 63.6 (63.5); H 6.7 (6.7); N 6.5 (6.7) %. IR: 1,606, 1,557 υ(C=O); 1,512 υ(C=C).
Cu(2-n-propylamino-4-i-propyltroponate)2 (19)
A green solid was obtained using (7) (0.50 g, 2.44 mmol) and copper acetate (0.24 g, 1.22 mmol). Yield 0.36 g, 62 %. Analysis for (19): Found [calc. for C26H36N2O2Cu]: C 66.9 (66.2); H 8.1 (7.6); N 5.7 (5.9). IR,: 1,591, 1,578 υ(C=O); 1,512 υ(C=C).
Synthesis of Cu(2-menthoxyethylaminotroponate)2 (20)
Using the methodology described earlier for 13–19 [17], (11) (0.36 g, 1.12 mmol) was reacted with copper(II) acetate (0.12 g, 0.60 mmol) to produce a dark green precipitate on standing (0.21 g, 27 %). Recrystallisation from hot acetone gave pale green crystals suitable for crystallographic analysis. Analysis for (20): Found [calc. C38H56O4N2Cu]: C 68.1 (68.3); H 8.4 (8.4); N 4.1 (4.2) %. IR: 1,592, 1,572 ν(C=O); 1,509 ν(C=C).
Synthesis of Cu(2-menthoxyethylamino-4-i-propyltroponate)2⋅H2O (21)
In a manner similar to that described for (20), (12) (1.00 g, 2.75 mmol) was reacted with copper(II) acetate (0.29 g, 1.45 mmol). A dark green precipitate was produced on standing. Crystals were obtained from hot toluene, but were too small for crystallographic analysis. Analysis for (21): Found [calc. C44H70O5N2Cu]: C 68.4 (68.6); H 9.0 (9.1); N 3.4 (3.6). IR: 3,200 ν(O–H); 1,595, 1,570 ν(C=O); 1,511 ν(C=C).
X-ray crystallography
Experimental details relating to the single-crystal X-ray crystallographic studies are summarised in Table 1. For all structures, data were collected on a Nonius Kappa CCD diffractometer at either 150(2) (5, 20) or 170(2) K (14) using Mo-Kα radiation (λ = 0.71073 Å). Structure solution followed by full-matrix least squares refinement was performed using the WinGX-1.70 suite of programmes [27], incorporating SHELXS and SHELXL within the SHELX-2013 suite of software [28]. Corrections for absorption (multi-scan) were made in all cases. Specific details: 14: the asymmetric is one-half of the molecule, and the remainder generated by an inversion centre located at the metal.
Antibacterial activity
Staphylococcus waneri was cultured in BHI broth (100 ml, Oxoid, UK) for 24 h at 37 °C (20 % CO2), transferred into centrifuge tubes and spun at 3,500 rpm for 7 min in a Mistral 1,000 centrifuge. The supernatant was decanted, and remaining pellet re-suspended in phosphate-buffered saline (PBS) (5 ml). This process was repeated twice, and the culture finally re-suspended in PBS (2 ml). The optical density at 610 nm (OD610) was then adjusted to 1.0 using PBS.
A known weight of compound was dissolved in DMSO to make a 10 % solution of the active formulation. 200 μL of this was then added to the bacterial suspension (190 μL) and left for 1 min. After decanting, the culture was rinsed 3 times with water (200 μL) and BHI broth (200 μL) was added followed by mineral oil (80μL), and the resultant incubated at 37 °C in a Dynatec plate reader. The absorbance at 610 nm was monitored every fifteen minutes for 18 h, and the time taken for culture regrowth to return OD610 to 0.5 noted.
Results and discussion
A range of 2-alkylaminotropones (1–7) were synthesised from a straightforward reaction between tosylated tropolone and the appropriate amine (Scheme 1). For those amines that are commercially available as aqueous solutions (RNH2, R = Me, Et), the reactions were carried out in CH2Cl2, as this facilitates separation of the product into the organic layer. For the remaining amines (R = C3H7, C4H9, C6H5CH2), the reactions were carried out in DMSO. In the cases of 6 and 7, tosylated hinokitiol was prepared as a mixture of isomers (a, b) in the ratio 9:1 [29], which leads to a mixture of isomeric 2-alkylamino-4-i-propyltropone products; no attempt to separate these isomers was made, and the mixture was used without further purification. However, this complication manifested itself in the purification of the alkylaminotropone ligands, which proved more challenging than for 1–5. In the cases of 6 and 7, while the ligand identity was confirmed by NMR, purification was effected by the formation of the copper complexes 18 and 19. Related 2-alkylamino-4-i-propyltropones where R = Et, n-Bu, Bz were impure as prepared, and while the purity of the resulting copper complexes was enhanced, satisfactory microanalytical data could not be obtained.

Protocols for the synthesis of 2-alkylaminotropone ligands
In addition to these unfunctionalised substituents, we have also prepared analogues containing the flavourant, menthol (Scheme 2). This follows a sequence in which menthyoxyacetic acid (8) is converted to the acetyl chloride and then the amide (9) using aqueous ammonia. 9 was then reduced to the amine (10), a colourless oil, with excess LiAlH4 [26] and which was in turn used in the protocols of Scheme 1 to afford the flavourant-functionalised 2-alkylaminotropones (11) and (12).

Synthetic sequence for the formation of menthol-flavoured 2-alkylaminotropone ligands
NMR data for 1–7 are unexceptional, but show the presence of the amine R′ along with resonances for the tropone ring in the correct ratios, while data for 11, 12 confirm the linking of the 2-menthoxyethylamine with the tropolone. Both these compounds, brown oils, were isolated as monohydrates in analytically pure form after extraction from the reaction mixture; data for the major isomer (note: Scheme 1) are given in the Experimental.
1–7, 11,12 all show ν(NH) and ν(C=O) in the ranges 3,293–3,307 and 1,629–1,637 cm−1, respectively, in their IR spectra. The structure of 5 has also been determined (Fig. 1) and is that of a N–H⋯O=C hydrogen-bonded dimer, similar to analogues with R′ = 2,6-i-Pr2C6H3 [30] and 4-F-C6H4 [31], though differing from the monomeric R′ = t-Bu [32], the latter structure presumably dictated by packing of the non-planar R′. Like the other reported dimers, the C=O [1.2573(18) Å] and C(2)-N(1) [1.3457(11) Å] in 5 suggest some lengthening with respect to the monomeric R′ = t-Bu derivative [1.242(4), 1.348(4) Å, respectively] as anticipated [32], though the large esds in these data preclude definitive comment. The C–C bonds around the ring between C(2)-C(7) [1.375(2)–1.398(2) Å] evidence delocalisation, while the two C–C(1) bonds are notably longer [C(1)-C(2) 1.485(2), C(1)-C(7) 1.432(2) Å]. The nitrogen atom shows a degree of planarity via its bond angles [115.2(13)°–125.09(12)°] implying delocalisation of the lone electron pair.

The asymmetric unit of 5 showing the labelling scheme used in the text; thermal ellipsoids are at the 50 % level. Selected geometric data: O(1)–C(1) 1.2573(18), N(1)–C(2) 1.3457(18), N(1)–C(8) 1.449(2), C(1)–C(7) 1.432(2), C(1)–C(2) 1.485(2), C(2)–C(3) 1.3982(19), C(3)–C(4) 1.398(2), C(4)–C(5) 1.375(2), C(5)–C(6) 1.398(2), C(6)–C(7) 1.376(2) Å, C(2)–N(1)-C(8) 125.09(12), C(2)–N(1)–H(1) 115.2(13), C(8)–N(1)–H(1) 119.3(13)°. Hydrogen bond: H(1)–O(1) 2.9601(16), N(1)⋯O(1) 2.19(2) Å, ∠N(1)–H(1)⋯O(1) 145.8(17)°. Symmetry operation: 1 − x, − y, − z
Copper derivatives (13–21) of the aminotropones described above were prepared by reaction of the above ligands with Cu(II) acetate in aqueous ethanol: 
The products are green solids, in which the IR ν(C=O) stretch has shifted 30–70 cm−1 to lower wavenumber, indicating C=O→Cu coordination. Furthermore, ν(NH) is absent in all these complexes, which, in the case of 21, a combination of microanalysis and a broad ν(OH) observed in the IR at 3,200 cm−1 suggest is a monohydrate.
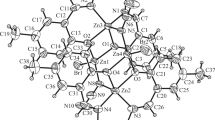
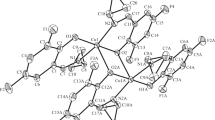
The structures of both 14 (Fig. 2) and 20 (Fig. 3) have been determined. The structures of both compounds adopt four-coordinate square-planar geometry as seen in the only other simple analogous copper compound reported to date (R′ = Me) [18], though examples with R′ = Et along with further functionalisation of the seven-membered ring [5-C5H4FeC5H5] also adopt this arrangement [19]. In the case of 14, the molecule is centrosymmetric about an inversion centre at the metal, while 20 lacks this symmetry by virtue of the flexible pendant group on nitrogen. The Cu–O and Cu–N bond lengths (Figure captions) are similar to those in 13 reported by others [18]. All three copper aminotropones show a general lengthening of the C=O by ca. 0.04 Å on complexation, while the NC(ring) bond contracts by ca. 0.02 Å, though in 20 these two C–N bonds are tending towards distinction [1.319(8), 1.340(9) Å], again reflecting the lower symmetry of the species; the nitrogen remains essentially planar in all cases. In the lattice, both 14 and 20 adopt a “slipped stack” arrangement with a π–π separation of ca. 4.2 Å.

The asymmetric unit of 14 showing the labelling scheme used in the text; thermal ellipsoids are at the 50 % level. Selected geometric data: Cu–O 1.9322(9), Cu–N 1.9366(10), O–C(3) 1.2945(15), NC(1) 1.4623(15), NC(4) 1.3250(15); O–Cu–O′ 180, O–Cu–N 82.35(4), NCu-N′ 180, O–Cu–N′ 97.65(4)°. Symmetry operation: 1 − x,1 − y,1 − z

The asymmetric unit of 20 showing the labelling scheme used in the text; thermal ellipsoids are at the 50 % level. Selected geometric data: Cu–O(1) 1.931(5), Cu–O(3) 1.923(5), Cu–N(1) 1.926(6), Cu–N(2) 1.929(6), O(1)-C(1) 1.304(9), O(3)–C(21) 1.297(8), N(1)–C(7) 1.317(8), N(2)–C(27) 1.340(9); O(1)–Cu–O(3) 179.7(2), O(1)–Cu–N(1) 82.3(2), O(1)–Cu–N(2) 96.8(2), O(3)–Cu–N(1) 98.0(2), O(3)–Cu–N(2) 82.9(2), N(1)–Cu–N(2) 178.6(3)°
The antibacterial activity of 13, 17, 20 and 21, along with data for the bis(ethylmaltol)copper complex [16] for comparison, is shown in Fig. 4. This single species biofilm assay (against Staphylococcus waneri) is an in vitro model of plaque inhibition effects. The results obtained from this test reflect the antibacterial activity of the complexes and are plotted as graphs of absorbance at 610 nm against time, which shows the delay in biofilm regrowth after treatment. From this, the time taken to return to an OD610 of 0.5 is noted; the longer the time for the bacteria to regrow after treatment, the more effective the agent is deemed to be. Figure 4 shows that all four 2-alkylaminotropones are more effective than the formulation in an commercial toothpaste, though none are as active as the copper derivative of ethylmaltol [16], a related but O,O-chelating ligand. Interestingly, the presence of the flavourant does not reduce the efficacy of the compounds; in fact, the activity is marginally enhanced for these two species over the other copper 2-alkylaminotropones.

Antibacterial activity of new copper 2-alkylaminotroponate complexes 13, 17, 20 and 21 along with bis(ethylmaltolato) copper(II) [16] and a commercial toothpaste (Signal) for comparison. The plot shows the time taken for a culture of Staphylococcus waneri to regrow to half its original size after treatment with the agent
Supplementary information
CCDC 990207–990209 contains the supplementary crystallographic data for this paper (Table 1). These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
References
Arima Y, Nakai Y, Hayakawa R, Nishino T (2003) J Antimicrob Chemother 51:113
Morita Y, Matsumura E, Okabe T, Shibata M, Sugiura M, Ohe T, Tsujibo H, Ishida N, Inamori Y (2003) Biol Pharm Bull 26:1487
Inamori Y, Sakagami Y, Morita Y, Shibata M, Sugiura M, Kumeda Y, Okabe T, Tsujibo H, Ishida N (2000) Biol Pharm Bull 23:995
Inamori Y, Tsujibo H, Ohishi H, Ishii F, Mizugaki M, Aso H, Ishida N (1993) Biol Pharm Bull 16:521
Nomiya K, Onodera K, Tsukagoshi K, Shimada K, Yoshizawa A, Tada-aki I, Sugie A, Tsuruta S, Sato R, Kasuga C (2009) Inorg Chim Acta 362:43
Irving RJ, Post ML, Povey DC (1973) J Chem Soc, Dalton Trans 2:697
Berg J-E, Pilotti A-M, Söderholm A-C, Karlsson B (1978) Acta Crystallog B34:3071
Griffith WP, Pumphrey CA, Skapski AC (1987) Polyhedron 6:891
Avdeef A, Costamanga JA, Fackler JPJ (1974) Inorg Chem 13:1854
Davis AR, Einstein FWB (1974) Inorg Chem 13:1880
Lockhart TP, Davidson F (1987) Organometallics 6:2471
Mori A, Yamamoto S-I, Takemoto M, Ujiie S (2002) Chem Lett 2:186
Mori A, Mori R, Takemoto M, Yamamoto S-I, Kuribayashi D, Uno K, Kubo K, Ujiie S (2005) J Mater Chem 15:3005
Creeth J, Molloy KC, Wright P (2000) Oral care compositions. Patent WO 00/16736
Barret MC, Mahon MF, Molloy KC, Wright P (2000) Main Group Met Chem 23:663
Barret MC, Mahon MF, Molloy KC, Steed JW, Wright P (2001) Inorg Chem 40:4384
Barret MC, Mahon MF, Molloy KC, Wright P, Creeth JE (2002) Polyhedron 21:1761
Steyl G, Muller TJ, Roodt A (2010) Acta Crystallog Sect E Struct Rep Online 66:m1508
Miyake Y, Watanabe S, Aono S, Nishinaga T, Miyazaki A, Enok T, Miyasaka H, Otani H, Iyoda M (2008) Chem Commun 44:6167
Nishinaga T, Aono T, Iomura E, Watanabe S, Miyake Y, Miyazaki A, Enoki T, Miyasaka H, Otani H, Iyoda M (2010) Dalton Trans 39:2293
Meyer N, Lohnwitz K, Zulys A, Roesky PW, Dochnahl M, Blechert S (2006) Organometallics 25:3730
Reddington MV (1998) J Chem Soc Perkin Trans II 143
Rasika-Dias HV, Jin W (1996) Inorg Chem 35:6546
Biggi G, Del Cima F, Pietra F (1973) J Am Chem Soc 95:7101
Cochran TG, Huitric AC (1971) J Org Chem 36:3046
Minardi G, Schenone P (1968) Farmaco Ed Sci 23:1181
Farrugia LJ (1999) J Appl Crystallogr 32:837
Sheldrick G (2008) Acta Cryst A64:112
Yanagisawa T, Kosakai K, Yasunami M, Takase K (1990) Chem Pharm Bull 38:3355
Hicks FA, Jenkins JC, Brookhart M (2003) Organometallics 22:3533
Steyl G (2007) Acta Crystallogr Sect E: Struct Rep Online 63:o4353
Siwatch RK, Kundu S, Kumar D, Nagendran S (2011) Organometallics 30:1998
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Barret, M.C., Bhatia, P.H., Kociok-Köhn, G. et al. New copper(II) 2-(alkylamino)troponates. Transition Met Chem 39, 543–551 (2014). https://doi.org/10.1007/s11243-014-9830-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-014-9830-0