
Abstract
The synthesis, structures and antibacterial studies of silver complexes of N-heterocyclic carbene (NHC) ligands are reported. The NHC precursors, 2,6-bis(3-allylimidazol-1-ylmethyl) pyridine hexafluorophosphate (1) and 2,6-bis(3-allylbenzimidazol-1-ylmethyl)pyridine dibromide (2) were treated with Ag2O to yield 2,6-bis(3-allylimidazol-1-ylmethyl)pyridine disilver bis(hexafluorophosphate) (3) and 2,6-bis(3-allylbenzimidazolium-1-ylmethyl)pyridine disilver bis(hexafluorophosphate) (4). All four compounds were characterized by physico-chemical and spectroscopic techniques and by single-crystal X-ray diffraction. The compounds were screened for their antibacterial activities against Staphylococcus aureus (ATCC 12600) as a Gram-positive and Escherichia coli (ATCC 11303) as a Gram-negative bacteria. Compounds 1 and 2 showed no inhibition while 3 and 4 inhibited the growth of these bacteria. The nuclease activities of the compounds were evaluated by gel electrophoresis, and the results indicate that complexes 3 and 4 can degrade both DNA and RNA.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
N-heterocyclic carbene (NHC) complexes of silver(I) are a well-studied class of organometallic compounds with relevance to organic, organometallic and carbene transfer chemistry [1]. The development of Ag(I)–NHC complexes and their applications are usually connected with the yield and cost of their synthesis [1]. Ag(I)–NHC complexes are easily prepared by reactions of azolium salts with silver bases like Ag2O, Ag2CO3 and AgOAc, and silver salts under basic phase transfer conditions [2–4]. Among these methods, in situ deprotonation of the azolium salts with silver bases is the most convenient and commonly used method. Ag–NHC complexes have proven to have interesting antimicrobial [5–8], antifungal [9] and anticancer [10, 11] properties.
Silver sulfadiazine and silver nitrate have been used widely as topical antimicrobial agents. However, they lose their effectiveness quickly, resulting in reinfection of the wound site. In addition, the development of drug resistance of some pathogenic bacteria to sulphonamides has reduced the use of some common antibiotics [12–14]. Hence, there is a need to develop novel silver compounds that can release silver ions gradually into the wound [15, 16]. To date, a large number of studies have been carried out on Ag(I)–NHC complexes as potential antimicrobial and anticancer agents, and their carbene transfer ability [17–21].
NHC metal complexes containing pincer-type ligands based on pyridine with several donor groups have been thoroughly explored [22–24], but the Ag(I)–carbene complexes with pincer-type ligands containing NHCs are relatively scarce compared to the monocarbene complexes [25, 26]. Imidazole-derived ligands based on bis(imidazolin-2-ylidene)-2,6-pyridine were first reported in 2000 [27], but it was in 2004 that the first pyridine-linked pincer Ag(I)–NHC complexes were reported, along with their antimicrobial activity [19]) Despite the encouraging early results, research on pincer-type NHC complexes has been focussed mostly on catalysis [28–31]. Our interest in pyridine-linked pincer silver NHC complexes is due to their ability to offer stability while retaining their activity in addition to the common electron-donating capabilities of the carbon [31]. This may lead to a degree of stabilization of the silver complexes, thereby controlling the release of Ag+ into the culture medium. Allyl substituents on such ligand precursors are capable of coordinating to the metal centre leading to a coordination environment made up of sp 2-carbon donors [32]. Inspired by this, we became interested in the coordination chemistry of N-heterocyclic carbenes bearing N-allyl substituents. We have previously reported that xylyl-linked bis-benzimidazolium salts and their respective dinuclear Ag–NHC complexes have potential activities against different types of cancer [33–36]. Therefore, compounds 1–4 were synthesized to further investigate this phenomenon in relation to their antimicrobial activities. Herein, we report the synthesis and crystal structures of Ag(I) benzimidazole and imidazole complexes stabilized by pyridine-based NHC ligands with allyl group as substituents. Their antimicrobial activities were investigated against Staphylococcus aureus as a Gram-positive and Escherichia coli as a Gram-negative bacterial strain.
Experimental
Materials and measurements
All chemicals were used as received. All solvents were redistilled except for acetonitrile and dimethyl sulfoxide, which were of AR grade. NMR spectra were recorded on a Bruker 500 MHz spectrometer at RT in DMSO-d 6 , using TMS as an internal standard. FTIR spectra were recorded on a Perkin-Elmer-2000 system spectrometer in the range 4,000–400 cm-1. Elemental analysis was carried out on a Perkin-Elmer series II, 2400 microanalyser. Melting points were measured using a Stuart Scientific SMP-1 (UK) instrument. The instruments are available at the School of Chemical Sciences, Universiti Sains Malaysia (USM). The X-ray single-crystal structure analysis was obtained using a Bruker smart ApexII-2009 CCD area detector diffractometer.
Synthesis of 2,6-bis(3-allylimidazol-1-ylmethyl)pyridine hexafluorophosphate (1)
A mixture of imidazole (1.00 g, 14.69 mmol) and KOH (1.23 g, 22.00 mmol) in DMSO (20 mL) was stirred at RT for 1 h. 2,6-Bis(bromomethyl)pyridine (1.95 g, 3.75 mmol) was then added portionwise, and the mixture stirred at RT for 2 h. The mixture was poured into water (200 mL) and extracted with chloroform (4 × 25 mL). The combined extract was filtered through four plies of Whatman filter papers in order to dry the extract. This process of filtration was repeated thrice to collect a clear solution of the desired compound, which was subsequently evaporated under reduced pressure. The desired compound, 2,6-bis(N-imidazole-1-ylmethyl)pyridine, was characterized by NMR and compared with the literature data before further use [37]. Next, a mixture of 2,6-bis(N-imidazole-1-ylmethyl)pyridine (0.50 g, 2.09 mmol) and allyl bromide (0.51 g, 4.18 mmol) in acetonitrile (35 mL) was refluxed at 80 °C for 20 h. The solvent was removed under reduced pressure to give 2,6-bis(3-allylimidazolium-1-yl methyl)pyridine dibromide as a yellowish oil, which was reacted with a solution of KPF6 (0.70 g, 3.80 mmol) in methanol (20 mL) to yield compound 1. Recrystallization from methanol gave the compound as colourless crystals. Yield: 0.68 g (76 %). Melting point: 166–167 °C. 1H NMR (500 MHz, DMSO-d 6 , 298 K, δ ppm): 4.88 (4H, d, J = 6.0 Hz, 2 N– × CH2); 5.31–5.43 (4H, d.d, J = 6.0, 5.5 Hz, 2 × CH2); 5.55 (4H, s, 2 × CH2-pyridine); 6.00–6.13 (2H, m, 2 × CH); 7.69 (4H, d, J = 6.0 Hz, imidazolium H4′, H5′); 7.49 (2H, d, J = 8.0 Hz pyridine-H); 7.97 (1H, t, J = 8.0 Hz, pyridine-H); 9.16 (2H, s, 2 × imidazolium H2′); 13C {1H} NMR (125 MHz, DMSO-d 6 , 298 K, δ ppm): 51.0 (N–CH2), 52.7 (CH2-pyridine);120.4 (–CH=CH2);142.6 (imidazole Ar–C); 136.1 (–CH = CH2) 138.9 (imidazolium-C2), 123.4, 134.7, 153.4 (pyridine Ar–C). FTIR (KBr disc) cm−1, ~2,966, 3,079 ν (C–H), 1,601 ν (C=N, imidazole), 1,578 ν (C=N, pyridine), 1,646 ν (C=C). Anal. Calc. for C19H23N5F12P2: C, 37.3; H, 3.8; C, 11.5. Found: C, 37.7; H, 4.0; N, 11.8.
Synthesis of 2,6-bis(3-allylbenzimidazol-1-ylmethyl)pyridine dibromide (2)
A mixture of benzimidazole (1.00 g, 8.46 mmol) and KOH (0.71 g, 12.68 mmol) in DMSO (20 mL) was stirred for 1 h at RT. 2,6-Bis(bromomethyl)pyridine (1.12 g, 4.23 mmol) was added portionwise; the mixture was stirred at RT for 2 h, then poured into water (200 mL) and cooled on ice; the resulting white precipitate was filtered off, washed with water (4 × 5 mL) and dried at 70 °C. The desired compound, 2,6-bis(N-benzimidazole-1-ylmethyl) pyridine, was characterized by NMR and compared with literature data before further use [37]. Next, a mixture of 2,6-bis(N-benzimidazole-1-ylmethyl)pyridine (0.72 g, 2.10 mmol) and allyl bromide (0.51 g, 4.20 mmol) in acetonitrile (35 mL) was refluxed at 80 °C for 20 h. The solvent was removed under reduced pressure to give 2,6-bis(3-allylbenzimidazolium-1-ylmethyl)pyridine dibromide as a white solid. Recrystallization from methanol gave the compound as colourless crystals. Yield: 0.65 g (74 %). Melting point: 177–178 °C. 1H NMR (500 MHz, DMSO-d 6 , 298 K, δ ppm): 5.15 (4H, d, J = 6.0 Hz, 2 × N–CH2); 5.38 (4H, d, J = 6.0 Hz, 2 × CH2); 5.41 (4H, s, 2 × CH2-pyridine); 6.00–6.13 (2H, m, 2 × CH); 7.43 (2H, t, J = 8.5 Hz, Ar–H); 7.55 (2H, d, J = 8.5 Hz, Ar–H); 7.66 (2H, t, J = 7.5 Hz, Ar–H); 7.94 (2H, d, J = 8.5 Hz, pyridine-H); 8.01 (1H, t, J = 8.5 Hz, pyridine-H) 9.83 (2H, s, 2 × benzimidazolium H2′); 13C {1H} NMR (125 MHz, DMSO-d 6 , 298 K, δ ppm): 48.9 (N–CH2), 50.6 (CH2-pyridine); 113.8 (–CH=CH2); 113.5, 120.6, 142.6 (benzimidazole Ar–C); 130.8 (–CH=CH2) 138.9 (benzimidazolium-C2), 122.5, 131.1, 153.0 (pyridine Ar–C). (KBr disc) cm−1, ~3,104, 3,159 ν (C–H), 1,596 ν (C=N, imidazole), 1,574 ν (C=N, pyridine), 1,664 ν (C=C); Anal. Calc. for C27H29N5Br2O: C, 54.1; H, 4.8; N, 11.7. Found: C, 54.6; H, 5.1; N, 11.9.
Synthesis of 2,6-bis(3-allylimidazol-1-ylmethyl)pyridine disilver(I) bis(hexafluorophosphate) (3)
An acetonitrile solution (30 mL) of 1 (0.50 g, 0.82 mmol) and Ag2O (0.38 g, 1.64 mmol) was stirred at 50–60 °C temperature for 24 h. The mixture was filtered through a pad of Celite, and the filtrate was slowly evaporated to precipitate a white solid. The solid was redissolved in acetonitrile, and diethyl ether was added to reprecipitate a solid, which was isolated by filtration and vacuum-dried to yield 3. Crystals suitable for X-ray diffraction were obtained by slow diffusion of the salt solution using diethyl ether and acetonitrile at ambient temperature. Yield: 0.53 g (76 %) Melting point: 222–224 °C. 1H NMR (500 MHz, DMSO-d 6 , 298 K, δ ppm): 5.11 (4H, d, J = 5.0 Hz, 2 × N–CH2); 5.16–5.25 (4H, d.d, J = 6.0, 5.5 Hz, 2 × CH2); 5.67 (4H, s, 2 × CH2-pyridine); 6.00–6.13 (2H, m, 2 × CH); 7.69 (4H, d, J = 6.0 Hz, imidazolium H4′, H5′); 7.73 (2H, d, J = 8.0 Hz, pyridine-H); 7.80 (1H, t, J = 8.0 Hz, pyridine-H); 13C {1H} NMR (125 MHz, DMSO-d 6 , 298 K, δ ppm): 53.2 (N–CH2), 55.5 (CH2-pyridine); 121.6 (–CH=CH2); 122.9 (–CH=CH2); 139.0 (imidazolium C4′and C5′); 138.9, 155.7 (pyridine Ar–C), 179.6 (imidazolium C2-Ag). (KBr disc) cm−1, ~3,104, 3,159 ν (C–H), 1,595 ν (C=N, imidazole), 1,578 ν (C=N, pyridine), 1,601 ν (C=C); Anal. Calc. for C38H42N10Ag2F12P2: C, 39.8; H, 3.7; N, 12.2. Found: C, 40.4; H, 4.1; N, 12.8.
Synthesis of 2,6-bis(3-allylbenzimidazol-1-ylmethyl)pyridine disilver(I) bis(hexafluorophosphate) (4)
This compound was prepared in a similar way 3, except that 1 was replaced with 2 (0.68 g, 0.96 mmol) and Ag2O (0.44 g, 1.92 mmol). Yield: 0.63 g (70 %). Melting point: 235–237 °C; 1H NMR (500 MHz, DMSO-d 6 , 298 K, δ ppm): 5.15 (4H, d, J = 6.0 Hz, 2 × N–CH2); 5.16–5.25 (4H, d,d, J = 6.0, 5.5 Hz, 2 × CH2); 5.67 (4H, s, 2 × CH2-pyridine); 6.00–6.13 (2H, m, 2 × CH); 7.18 (2H, t, J = 8.5 Hz, Ar–H); 7.29 (2H, d, J = 8.5 Hz, Ar–H); 7.40 (2H, t, J = 7.5 Hz, Ar–H); 7.35 (2H, d, J = 8.5 Hz, pyridine-H); 7.75 (1H, t, J = 8.5 Hz pyridine-H); 13C {1H} NMR (125 MHz, DMSO-d 6 , 298 K, δ ppm):): 50.7 (N-CH2), 52.9 (CH2-pyridine); 111.8 (–CH=CH2); 112.1, 118.2, 138.5 (benzimidazole Ar–C); 133.0 (–CH=CH2) 121.2, 133.4, 154.9 (pyridine Ar–C), 171.6 (benzimidazolium C2-Ag). (KBr disc) cm−1, ~3,104, 3,159 ν (C–H), 1,596 ν (C=N, imidazole), 1,570 ν (C=N, pyridine), 1,664 ν (C=C) Anal. Calc. for C54H51N10Ag2F12P2: C, 48.2; H, 3.8; N, 10.4. Found: C, 48.5; H, 4.1; N, 10.8.
Antibacterial studies
Stock solutions of all compounds were prepared using DMSO. Antimicrobial tests were performed using the Kirby–Bauer disc diffusion method [38]. Single colonies of E. coli (ATCC 11303) and S. aureus (ATCC 12600) from fresh culture agar plates were, respectively, cultured in two bottles containing nutrient broth solution and incubated overnight at 37 °C. The turbidity of each culture was adjusted by comparing it to 0.5 McFarland standard, which is equal to 1.58 × 108 CFU/mL or 0.5 (O.D600 reading). Using sterile cotton buds, each bacterial lawn culture was spread uniformly on different agar plates before placing the antimicrobial assay discs on the plate. Four discs were placed on the agar plate, and 5 μL volumes of the compounds was loaded on the discs with concentrations at 100 and 50 μg/mL. The plates were incubated at 37 °C for 24 h, and the diameters of the inhibition zones were measured in millimetre. The effectiveness of these compounds in relation to inhibition zone was compared to silver nitrate, based on its established antimicrobial properties [39], and streptomycin that were used as positive controls. The minimum inhibitory concentration (MIC) of each compound was determined based on the lowest concentration of the compound that inhibited the growth of bacteria using the broth dilution method [40]. Single colonies of S. aureus and E. coli were isolated from agar plates and were grown in 5.0 mL LB broth. The solutions were incubated at 37 °C and shaken at 180 rpm overnight to yield bacteria solutions. Stock solutions of the compounds were prepared by dissolving each in DMSO to prepare stock concentrations at 50 mg/mL. From each stock solution, 2 μL of the compounds, AgNO3 and streptomycin, was dissolved in 2 mL of the broth culture and used to prepare four serial dilutions of 75, 50, 25 and 12.5 μg/mL, respectively. Serial dilutions were done for each compound by transferring 0.5 mL (75 μg/mL) of the compound solution from the first tube to the second tube already containing 1 mL of nutrient broth. Next, 1 mL from tube 2 was again transferred into tube 3, and same was repeated for tube 4 to give concentrations of 50, 25 and 12.5 μg/mL, respectively. Prepared bacteria solution (5 μL) was added to each tube, and the tubes were incubated at 37 °C for 16 h in a shaking incubator at 180 rpm. Bacteria growth was noted by turbidity of the solution in the tubes, and MIC was determined by the lowest concentration lacking turbidity.
Gel electrophoresis
The electrophoresis method was employed to study the efficiency of cleavage by the test compounds [41]. The extracted plasmid DNA and RNA, pTS414 (10 μg/mL), and 1 μL of the test sample (50 μg/mL) were mixed in 50 mM Tris–HCl buffer (pH 8.00). The contents were incubated for 8 h at 37 °C. Plasmid extraction was done using a plasmid purification kit (Intron biotechnology, Korea) without the addition of RNase in order to extract both RNA and DNA. The electrophoresis was performed using 0.8 % agarose gel. For each sample, 5 μL of the mixture was loaded into the well. The voltage used was 90 V running on 0.5 × Tris–Acetate EDTA (TAE) buffer, and the gel was stained with ethidium bromide solution (10 μg/mL) for 15 min. The gel was subsequently exposed to UV light and captured by a gel documentation system (FluorChem HD2; Cell Bioscience). The nucleating ability of the synthesized imidazolium and benzimidazolium salts/Ag complexes (1–4) was determined by its efficiency in cleaving/degrading the plasmid DNA and RNA.
Results and discussion
Synthesis
The bis(imidazolium/benzimidazolium) salts possessing pyridine spacers show an interesting pincer-type ligand framework, which gives an additional N-donor site that is capable of developing a stable backbone scaffold for increased stability and reactivity. The imidazolium and benzimidazolium salts 1 and 2 were prepared from imidazole and benzimidazole, respectively, by stepwise alkylation with 2,6-bis(bromomethyl)pyridine in the presence of KOH in DMSO at room temperature for 2 h. The reactants were subsequently converted into their respective quaternary salts by the reaction with allyl bromide, followed by treatment with KPF6 to obtain stabilized pincer-type NHC salts (Scheme 1). The salts 1 and 2 exhibit good solubility in polar organic solvents such as CH2Cl2, CH3CN and DMSO, but are insoluble in dioxane, ethyl acetate, diethyl ether and hexane. The enhanced acidity of the benzimidazole and imidazole H2 is a primary factor in the synthesis of Ag(I)–NHC complexes [42, 43]. The Ag(I)–NHC complexes 3 and 4 were prepared via the reaction of the ligand precursors 1 and 2, respectively, with Ag2O in 1:2 ratio in acetonitrile at 50–60 °C for 24 h.

Synthesis of bis(imidazolium/benzimidazolium) salts and their respective dinuclear Ag(I)–NHC complexes
Spectroscopy studies
Solution studies of these complexes were essential in order to identify the bioactive species resulting from dissolution of the solid complex and so to understand the underlying biochemical mechanism involved in the antibacterial activity. This study is also relevant to the antibacterial studies since the complexes have to be maintained in culture medium for at least 24 h. Therefore, we have determined the stabilities of the Ag-NHC complexes 3 and 4 in both 10 and 100 % DMSO-d 6 aqueous solution. Both complexes are stable in aqueous solution for 24 h, since their 1H NMR spectra remained unchanged after 24 h. The 1H NMR spectrum of complex 3 after 24 h in 10 % aqueous DMSO is shown in Fig. 1 and is identical to the spectrum obtained after 30 min (see supplementary data).

Representative portion of the 1H NMR spectrum of complex 3 after 24 h
In order to further assess the stability of the complexes in the broth mixture, 10 mg/mL stock solutions of 3 and 4 in DMSO was prepared and added in a 1:1 ratio to LB broth prepared in DMSO. This was done to imitate the conditions of the MIC evaluation experiments. The 1H NMR and 13C NMR spectra were taken after 15 min and 48 h. Both complexes demonstrated stability in the LB/DMSO broth mixture at 0 °C, room temperature and 37 °C, as shown by the unchanged 1H and 13C NMR spectra after 48 h.
The FTIR spectra of the carbene precursors show a band of medium intensity at around 2,928–3,431 cm−1, which is assigned to ν(C–H) (both aliphatic and aromatic) in benzimidazolium and imidazolium salts. The spectra of the salts show a band of medium intensity at around 1,561 cm−1 for 1 and 1,595 cm−1 for 2 assigned to the pyridine ring vibrations. Complexation of these NHCs with silver alters their spectra, which is evident in range of 1,200–1,610 cm−1, assignable to imidazole ring ν(C=N) vibrations [44]. These signals have a noticeable shift of about 35 cm−1 for 3 and 60 cm−1 for 4, arising from the rigid pyridine moiety. A moderate intensity band observed in the range 1,601–1,664 cm−1 is ascribed to the stretching vibration (C=C) of the allyl functionality in the carbene precursors [45], and this remains unchanged in the carbene complex spectra. This is an indication that the allyl functionality is outside the coordination sphere of the coordinatively saturated Ag(I) centre [46]. The NHC carbon upon complexation with silver undergoes spectroscopic changes, this is observed in the range 1,190–1,570 cm−1, and a characteristic “four fingers (f.fs)” pattern was observed for both 3 and 4. This pattern is quite different from the spectra of corresponding benzimidazolium and imidazolium salts 1 and 2, which has been previously reported by our research group [47].
The 1H NMR spectra of compounds 1–4 were in accordance with the expected structures. In the 1H NMR spectra of 3 and 4, the absence of resonances for the benzimidazolium and imidazolium carbene protons (NCHN) confirms the formation of the expected Ag(I)–NHC complexes [48]. Likewise, the 13C NMR signals for the carbene carbon (NCN) were observed downfield as expected for Ag(I)–NHC complexes [49]. The benzimidazolium and imidazolium C2-H groups show characteristic signals in the 1H NMR spectra of 1 and 2 in the range δ 9.16–9.83 ppm. Three typical signals due to the allyl protons were observed in the range of δ 4.88–5.43 ppm, and the presence of sharp singlets and multiplets at δ 6.0 and δ 7.49–8.01 ppm in all spectra were assigned to the pyridyl and aromatic protons. Apart from the absence of the benzimidazolium and imidazolium protons, no other significant changes were observed in the spectra of the complexes compared to the free ligands. In the 13C NMR spectra of complexes 3 and 4, the signals for the carbene carbon were observed at δ 171.61 and 179.61 ppm, respectively.
Crystal structures
The pro-ligand 1 crystallized in the monoclinic space group C2/c (see Table 1 for the crystal data details) with the asymmetric unit consisting of one 2,6-bis(3-allylimidazolium-1-ylmethyl)pyridine cation and two hexafluorophosphate anions (Fig. 2). The central pyridyl ring makes dihedral angles of 110.22(13)° and 114.46(14)° with the 1H-imidazol-3-ium ring and the minor components of the disordered 1H-imidazol-3-ium ring. The internal ring angles of the imidazole ring, N1-C1-N2 and N4-C13-N5 at the two carbene centres are 108.35(8)° and 108.49(10)°, respectively (Table 2). The pyridine rings in adjacent molecules are observed to form intermolecular π–π interactions, with the distance between the two centroids of the adjacent pyridyl rings being 3.507(5) Å (see supplementary data).

Molecular structure of 1 with thermal ellipsoids shown at 50 % probability. Hexafluorophosphate anions in the lattice and hydrogen atoms are omitted for clarity
Compound 2 crystallizes in the triclinic space P − 1 with 2,6-bis(3-allylimidazolium-1-ylmethyl)pyridine, two bromide anions and one water molecule contained in the asymmetric unit (Fig. 3). The benzimidazolyl units in 2 are on either side of the central pyridine core, with a dihedral angle of 125.37(17)°. Internal ring angles of the imidazole moiety, N1-C1-N2 and N4-C18-N5 at the two carbene centres are 110.29(17)° and 110.94(15)°, respectively. The two benzimidazole units and the central pyridine core form a z shape and are also perpendicular to the plane of the central pyridine unit. Face-to-face π–π interactions are observed between benzimidazole rings (back to back), with the distances between the centroids of the aromatic rings ranging from 3.522 to 3.544 Å, giving rise to the formation of a 1D supramolecular network (see supplementary data). One of the bromide ions acts as a bifurcated hydrogen bond acceptor to adjacent water molecules in the lattice. The formation of C–H···Br hydrogen bonds in the structure helps to stabilize the crystals.

Molecular structure of 2 with thermal ellipsoids shown at 50 % probability. Hydrogen atoms, bromide ions and water molecule in the lattice are omitted for clarity
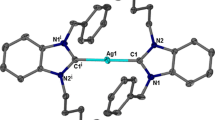
Complex 3 crystallizes in the monoclinic space group P21/c with half of the molecule in the asymmetric unit. Two silver ions are interposed between two units of 1, in which each ligand displays a μ-κ1(C)Ag:κ1(C′)Ag′ coordination mode, binding the Ag(I) through the imidazole carbene carbons to give an overall dinuclear complex structure (Fig. 4). The presence of two hexafluorophosphates in the lattice balances the charge on the complex. The dihedral and internal ring angles of 3 are about 15–16° and 6–7° smaller when compared with the salt of the free ligand. This could be ascribed to the fact that the NHC carbon atom, upon complexation with Ag(I), donates charge density to the metal cation, resulting in modulation of the ring shape. The Ag(I) ions in complex 3 display a linear coordination geometry, having the C1-Ag1-C14i (and symmetry equivalent) angle at 171.19(14)°. The Ag-C bond distances for both sets of imidazolium rings, Ag1-C1 and Ag1-C14i which are 2.096(8) and 2.094(12) Å, respectively, are in close accordance with other comparable imidazolium complexes of Ag(I) [50]. The separation between the two Ag(I) ions in this dinuclear complex is 4.784(5) Å respectively (Table 3).

Molecular structure of 3 with thermal ellipsoids shown at 50 % probability. Hydrogen atom and molecules in the lattice are omitted for clarity. Symmetry elements used: i=2 − x, 1 − y, z
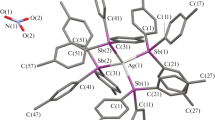
Complex 4 crystallizes in the orthorhombic space group P212121 with the entire molecule in the asymmetric unit. This dinuclear complex is constructed from two ligands of 2 that sandwich two silver cations through the NHC carbon atoms, while two hexafluorophosphate anions in the lattice neutralize the cationic molecule (Fig. 5a). The benzimidazolyl units lie on either side of the central pyridine core with a dihedral angle of 111.5(6)°. The N–C and P–F bond distances are in the range of 1.333(11)–1.488(9) and 1.575(6)–1.612(5) Å, respectively. The internal ring angles of benzimidazole (N–C–N) at the carbene centre are 106.57(7)° for N1–C1–N2 and 105.31(9)° for N6–C28–N7 (Table 4). The separation between Ag(I) ions is 4.468(5) Å. The bond angle at the carbene carbon contracts from 110.31° to 104.1°, and these internal ring angles are 4°–5° smaller relative to the angles in the free ligand. This is also in line with the shifts in charge density from the NHC carbon to the metal, which is responsible for the observed deviation in ring shape. There are π–π interactions between phenyl rings in the benzimidazole moieties that link adjacent complexes with a distance of 3.663 Å, leading to the formation of a 1D stepped chain network (Fig. 5b).

a Molecular structure of 4 with thermal ellipsoids shown at 50 % probability. Molecules in the lattice and hydrogen atoms are omitted for clarity. b The stepped chain section of face-to-face π–π stacking. Symmetry elements used: i=x − 1, y, z; ii=1 + x, y, z
Antibacterial activities
Stock solutions of all the compounds were prepared in DMSO. All dilutions were carried out with distilled water. The concentrations of the test compounds were 100, 75, 50, 25 and 12.5 μg/mL, using silver nitrate for positive control and streptomycin as the standard drug. Compounds were screened for their antibacterial activity against E. coli and S. aureus using the disc diffusion method, and the MIC was determined based on the lowest concentration that inhibited the growth of the bacteria.
A comparative study of the ligand precursors and their complexes (inhibition zone in mm in Table 5) indicates that the complexes exhibited good inhibition against both the Gram-negative and Gram-positive bacteria, while their corresponding free ligands (1 and 2) showed no inhibition. Although there is evidence that some imidazolium salts can display antimicrobial activity [51, 52], the present results are consistent with studies on other reported ligand precursors in the literature [53–55]. Both of the new complexes were effective at inhibiting the growth of Gram-positive and Gram-negative bacteria, with MIC values between 75 and 25 μg/mL for 3 and 4, respectively. These values are comparable to the MIC for silver nitrate (50 μg/mL) and some other related results in the literature [53–55], showing that these new Ag–NHC complexes are quite potent. The MIC values are summarized in Table 6. Generally, the complexes show greater antibacterial activities when compared with the ligand precursors, which is in general agreement with the literature [56].
Nuclease activities
Gel electrophoresis was used to evaluate the nuclease properties of compounds 1–4. An agarose gel image showing the nuclease activity of the compounds is shown in Fig. 6. Compounds were studied for their nuclease activity against plasmid pTS414 DNA/RNA. The imidazolium salts 1 and 2 displayed no visible activity towards either DNA or RNA degradation (Lane 3 and 4). However, their respective complexes 3 and 4 displayed distinct nuclease activity, especially for complex 3, which clearly degraded the nucleic acids as can be seen in lane 5. The plasmid DNA was totally degraded, leading to the loss of the associated bands on the gel [57]. The appearance of smearing in lane 6 reveals some degradation of the nucleic acids. The control DNA alone (Lane 2) does not show any apparent cleavage. Complexes 3 and 4 probably have similar pathways of action, as they both show nuclease activity against both DNA and RNA.

Nuclease activity; Lane 1 marker; Lane 2 DNA/RNA alone; Lane 3 DNA/RNA + sample 1; Lane 4 DNA/RNA + sample 2; Lane 5 DNA/RNA + sample 3; Lane 6 DNA/RNA + sample 4
Conclusions
The two silver NHC complexes reported here both showed good-to-moderate activity against both Gram-negative and Gram-positive bacteria; however, the benzimidazole-based compound (4) showed better activity than the imidazole-based compound (3). Both complexes also showed nuclease activity. Considering their stability in solution which may enhance the slow release of Ag+, we may conclude that the results of this work are promising, and further work is currently under way to improve these results by varying the substituents on the imidazole and benzimidazole rings in order to improve the biological activity. This should lead to enhanced solubility, stability and activity in biological media.
Supplementary Material
Crystallographic data for the structure in this work have been deposited at the Cambridge Crystallographic Data Centre, CCDC 951179, 939065, 945699 and 941463. Copies of these materials can be obtained from the director, CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: +44-1223-336033; e-mail: deposit@ccdc.cam.ac.uk/deposit.
References
Deblock MC, Panzner MJ, Tessier CA, Cannon CL, Youngs WJ (2011) RSC Catal Ser 6:119–133
Wang HMJ, Lin IJB (1998) Organometallics 17:972–975
Lee CK, Lee KM, Lin IJB (2002) Organometallics 21:10–12
Lin IJB, Vasam CS (2007) Coord Chem Rev 251:642–670
Özdemir I, Gürbüz N, Doğan O, Günal S, Özdemir I (2010) Appl Organomet Chem 24:758–762
Yığıt B, Gök Y, Özdemır I, Günal S (2012) J Coord Chem 65:371–379
Li Y, Dong X, Gou Y, Jiang Z, Zhu H-L (2011) J Coord Chem 64:1663–1669
Özdemir I, Demir S, Günal S, Arıcı C, Ülkü D (2010) Inorg Chim Acta 363:3803–3808
Nomiya K, Takahashi S, Noguchi R, Nemoto S, Takayama T, Oda M (2000) Inorg Chem 39:3301–3311
Liu W, Gust R (2013) Chem Soc Rev 42:755–773
Tan SJ, Yan YK, Lee PP, Lim KH (2010) Future Med Chem 2:1591–1608
Modak SM, Stanford JW, Bradshaw W, Fox CLJ (1983) Panminerva Med 25:181–189
Pirnay JP, Vos DD, Cochez C, Bilocq F, Pirson J, Struelens M, Duinslaeger L, Cornelis P, Zizi M, Vanderkelen A (2003) J Clin Microbiol 41:1192–2202
Modak SM, Fox CLJ (1985) J Trauma 25:27–31
Lansdown ABG, Sampson B, Laupattarakasem P, Vuttivirojana A, Dermatol Br J (1997) 137:728–735
Drousou A, Falabella A, Kirsner RS (2003) Wounds 15:149–166
Budagumpi S, Haque RA, Salman AW (2012) Coord Chem Rev 256:1787–1830
Papini G, Bandoli G, Dolmella A, Lobbia GG, Pellei M, Santini C (2008) Inorg Chem Commun 11:1103–1106
Melaiye A, Simons RS, Milsted A, Pingitore F, Wesdemiotis C, Tessier CA, Youngs WJ (2004) J Med Chem 47:973–977
Kascatan Nebioglu A, Melaiye A, Hindi K, Durmus S, Panzner M, Hogue L, Mallett R, Hovis C, Coughenour M, Crosby S, Milsted A, Ely D, Tessier C, Cannon C, Youngs WJ (2006) Med Chem 49:6811–6818
Ray S, Mohan R, Singh JK, Samantaray MK, Shaikh MM, Panda D, Ghosh PJ (2007) J Am Chem Soc 129:15042–15053
Hindi K, Siciliano T, Durmus S, Panzner M, Medvetz D, Reddy V, Hogue L, Hovis CE, Hilliard J, Mallett R, Tessier C, Cannon C, Youngs W (2008) J Med Chem 51:1577–1583
Poyatos M, Mata JA, Peris E (2009) Chem Rev 109:3677–3708
Peris E, Crabtree RH (2004) Coord Chem Rev 248:2239–2246
Garrison JC, Young WJ (2005) Chem Rev 105:3978–4008
Haque RA, Ghdhayeb MZ, Salman AW, Budagumpi S, Ahamed MBK, Abdul Majid AM (2012) Inorg Chem Commun 22:113–119
Chen JCC, Lin IJB (2000) J Chem Soc Dalton Trans 6:839–840
Wang Xin, Liu Shuang, Weng Lin-Hong, Jin Guo-Xin (2006) Organometallics 25:3565–3569
Normand AT, Cavell KJ (2008) Eur J Inorg Chem 14:2781–2800
John A, Ghosh P (2010) Dalton Trans 39:7183–7206
Kühl O (2007) Chem Soc Rev 36:592–607
Hahn FE, Holtgrewe C, Pape T (2004) Z. Naturforsch 59b:1051–1053
Haque RA, Iqbal MA, Khadeer Ahamed MB, Abdul Majeed AMS, Abdul Hameed Z (2012) Chem Cent J 6:68–71
Haque RA, Ghdhayeb MZ, Budagumpi S, Salman AW, Khadeer Ahamed MB, Abdul Majid AMS (2013) Inorg Chim Acta 394:519–525
Haque RA, Iqbal MA, Budagumpi S, Khadeer Ahamed MB, Abdul Majid AMS, Hasanudin N (2013) Appl Organomet Chem 27:214–223
Haque RA, Salman AW, Budagumpi S, Amirul AA, Abdul Majid AMS (2013) Metallomics 5:760–769
Jenkins DM, Betley TA, Peters JC (2002) J Am Chem Soc 124:11238–11239
Drew WL, Barry AL, O’Toole R, Sherris JC (1972) Appl Microbiol 24:240–247
Lansdown AB, Williams A, Chandler S, Benfield S (2005) J Wound Care 14:155–160
Wiegand I, Hilpert K, Hancock EWR (2008) Nat Protoc 3:163–175
Ramana N, Jeyamurugana R, Sakthivel A, Mitub L (2010) Spectrochim Acta A 75:88–97
Haque RA, Ahmed SA, Zetty ZH, Hemamalinic M, Fun HK (2011) Acta Crystallogr Sect E 67:3462
Baker MV, Brown DH, Haque AR, Simpson PV, Skelton BW, White AH, Williams CC (2009) Organometallics 28:3793–3803
Budagumpi S, Revankar VK (2010) Transit Met Chem 35:649–658
Wylie WNO, Lough AJ, Morris RH (2010) Organometallics 29:570–581
Liu OX, Li HL, Zhao XJ, Ge XX, Shi MC, Shen G, Zang Y, Wang XG (2011) Inorg Chim Acta 376:437–442
Haque RA, Iqbal MA, Asekunowo P, Abdul Majid AMS, Khadeer Ahamed MB, Umar MI, Al-Rawi SS, Al-Suede FSR (2013) Med Chem Res. doi:10.1007/s00044-012-0461-8
Baker MV, Brown DH, Haque RA, Skelton BW, White AH (2009) J Incl Phenom Macrocycl Chem 65:97–109
Wan XJ, Xu FB, Li QS, Song HB, Zhang ZZ (2005) Organometallics 24:6066–6068
Cambridge Structural Database, version 5.33; November 2011
Pernak J, Sobaszkiewicz K, Foksowicz-Flaczyk J (2004) Chem Eur J 10:3579–3585
Pernak J, Kalewska J, Ksycinska H, Cybulski J (2001) Eur J Med Chem 36:899–907
Murthy YL, Durga G, Jha A (2013) Med Chem Res 22:2266–2272
Ozdemir I, Ozcan EO, Giinal S, Gurbus N (2010) Molecules 15:2499–2508
Knapp AR, Panzner MJ, Medvetz AD, Wright BD, Tessier CA, Youngs WJ (2010) Inorg Chim Acta 364:125–131
Agwara MO, Ndifon PT, Ndikontan MK, Atamba MA (2008) Res J Chem Environ 12:87–92
Raman N, Baskaran T, Selva A (2008) J Iran Chem Res 1:29–139
Acknowledgments
The authors thank Universiti Sains Malaysia (USM) for the Research University (RU) Grant 1001/PKIMIA/811157. The authors are grateful to Prof A. A. Amirul of the School of biological Sciences, Universiti Sains Malaysia, for his useful technical assistance.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Haque, R.A., Asekunowo, P.O. & Razali, M.R. Dinuclear silver(I)-N-heterocyclic carbene complexes of N-allyl substituted (benz)imidazol-2-ylidenes with pyridine spacers: synthesis, crystal structures, nuclease and antibacterial studies. Transition Met Chem 39, 281–290 (2014). https://doi.org/10.1007/s11243-014-9801-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-014-9801-5