
Abstract
The complexes [(dppp)Pt{(μ-C2C5H4N)W(CO)4(PR3)}2] (dppp = Ph2P(CH2)3PPh2; PR3 = PPh3 (1), P(4-XC6H4)3 (X = Me (2), OMe (3), F (4)) P(NMe2)3 (5)) were prepared from [(dppp)Pt(C2C5H4N)2] and cis-[W(CO)4(PR3)(CH3CN)] and were characterized by 1H, 13C, 15N, 31P, 183W and 195Pt NMR spectroscopy. The complex [(Ph3P)2Rh(H)2(μ-PTC)W(CO)4(P(4-MeC6H4)3)] (6) was prepared similarly, from [(Ph3P)2Rh(H)2(PTC)] (thiazole nitrogen and carboxylate oxygen attached to the metal) and cis-[W(CO)4(P(4-MeC6H4)3)(CH3CN)] and was characterized by NMR (1H, 13C, 15N, 31P, 103Rh and 183W) and by X-ray crystallography, which shows π–π interactions between the bridging pyridyl and a phenyl group from a phosphine on each metal. In complexes 1–5, there is little variation in the platinum chemical shift, indicating that electronic influences through the pyridylacetylide bridge are minimal.
Graphical Abstract
A bimetallic complex of rhodium and tungsten bridged by 2-(4-pyridyl)-thiazole-4-carboxylate is reported together with an NMR spectroscopic study of a series of trinuclear complexes of platinum and tungsten bridged by 4-pyridylacetylide.

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Heteronuclear bi- and trimetallic complexes, in which the metals are connected by acetylide or oligoacetylide bridging ligands are of interest in the development of structurally organized materials in which the different metals display cooperative effects and communicative abilities [1–3]. A number of complexes of this type have 4-pyridylacetylide as the bridging ligand and are of the form [MC2(4-C5H4N)M′] (M = Ru, Rh; M′ = W, Re, Pd) [4–9] and [M{C2(4-C5H4N)M′}2] (M = Pd, Pt; M′ = Pt, Pd) [10–13]. The use of compounds such as these for the development of molecular wires, in which acetylide chains terminated at one or both ends by polypyridyl groups bridge two metal atoms, is of interest not only for electron transfer [14–16], but also for photoactive effects [17, 18]. Other types of bridging ligand that make use of a pyridyl group for attachment to a metal atom are 4-(4-pyrazolyl)pyridine [19] and 3,5-bis(2-pyridyl)-1,2,4-triazole [20]. We report complexes of 4-pyridylacetylide bridging platinum and tungsten and 2-(4-pyridyl)thiazole-4-carboxylate bridging rhodium and tungsten.
Experimental
THF was distilled from sodium/benzophenone, dichloromethane was distilled from P2O5, acetonitrile was dried over calcium hydride; other materials were of the highest available purity and were used without further treatment. [(dppp)Pt(4-C2C5H4N)2] was prepared by the method of Anderson and co-workers [21], [W(CO)4(phosphine)(CH3CN)] was prepared by the method of Adams and Perrin [22] with slight modification (solvent 100 % CH3CN, temperature 80 °C, Me3NO 1.2 equivalents added over 10 min, stirred 40 min) and [Rh(H)(PPh3)4] was prepared by the method of Robinson and co-workers [23]. All operations were performed under an argon atmosphere. Infrared spectra were recorded on a Bruker Tensor 27 FT-IR spectrometer; NMR spectra were recorded on a Bruker DRX 400 spectrometer operating at 400.13 MHz (1H), 100.62 MHz (13C), 40.54 MHz (15N) 161.98 MHz (31P), 12.65 MHz (103Rh), 16.67 MHz (183W) and 86.02 MHz (195Pt). Two-dimensional 15N–31P, 103Rh–31P, 183W–31P and 195Pt–31P spectra were obtained using the HMBC pulse sequence of Bax and co-workers [24]. Spectra were referenced to the generally accepted standards of TMS (1H, 13C), nitromethane (ext. standard) (15N), 85 % H3PO4 (ext. standard) (31P), Ξ = 3.16 MHz (103Rh) and aqueous Na2PtCl6 (0.220 M) (195Pt) and the recently proposed standard of Ξ = 4.15 MHz for 183W [25]. The rhodium complexes were found to have low solubility in commonly used solvents, necessitating the use of a solvent mixture, CD3OD/THF (1:1), for NMR spectroscopy.
X-ray structure determination
Intensity data were collected on a Bruker APEX II CCD area detector diffractometer with graphite monochromated Mo Kα radiation (50 kV, 30 mA) using the APEX 2 [26] data collection software. The collection method involved ω-scans of width 0.5° and 512 × 512 bit data frames. Data reduction was carried out using the program SAINT+ [27], and face-indexed absorption corrections were made using the program XPREP [27]. The crystal structure was solved by direct methods using SHELXTL [28]. Non-hydrogen atoms were first refined isotropically followed by anisotropic refinement by full matrix least-squares calculations based on F 2 using SHELXTL. Hydrogen atoms were first located in the difference map, then positioned geometrically and allowed to ride on their respective parent atoms. Crystal and structure refinement data for 6 are given in Table 1 and selected bond distances and angles in Table 2.
Preparation of [Pt(dppp){(μ-C2C5H4N)W(CO)4(PPh3)}2] (1)
A stirred solution of [Pt(dppp)(4-ethynylpyridine)2] (0.030 g, 0.037 mmol) and [W(CO)4(PPh3)(CH3CN)] (0.046 mg, 0.077 mmol) in THF (4 ml) was allowed to stand at room temperature for 16 h. The solvent was removed under vacuum leaving orange flakes which were thoroughly washed with diethyl ether. Yield 0.068 g (96 %). IR (compressed solid) ν(CC) 2116, ν(CO) 2007, 1864, 1829; 1H NMR (C6D6 300 K) δ 7.96 (mult, 2H, py 1,6), 7.68–7.45 (mults, 20H, Ph), 7.05–6.85 (mults, 30H, Ph), 6.02 (mult, 2H, py 3,5), 1.81 (mult, 4H, CH2P), 1.35 (mult, 2H, CH2); 13C NMR (C6D6 300 K) δ 210.14 (d (J(P,C) 5.1) CO), 209.25 (d (J(P,C) 32.9) CO), 205.60 (d (J(P,C) 7.1) CO), 154.50 (py 2,6), 135.16 (d (J(P,C) 36.2) PhPW 1), 133.70 (d (J(P,C) 11.7) PhPW 2,6), 131.1 (PhPPt 4), 129.67 (d (J(P,C) 1.9) PhPW 4), 128.52 (d (J(P,C) 9.2), 128.4–127.7 (signals overlapped by C6D6 signal, PhPPt), 126.45 (py 3,5), 121.93 (dd, (J(P,C) 146, 23) PtC), 107.36 (dd, Pt sat. (J(P,C) 18, 17; J(Pt,C) 300) Pt CC), 25.44 (mult, CH2P), 19.71 (CH 2 CH2P). 15N, 31P, 183W and 195Pt NMR data are given in Table 3.
Preparation of [Pt(dppp){(μ-C2C5H4N)W(CO)4(PR3)}2] (PR3 = P(4-RC6H4)3, R = Me (2), MeO (3), F (4); P(NMe2)3 (5))
These compounds were prepared in the same manner as 1. IR (compressed solid) ν(CC) 2116, ν(CO) 2006, 1868, 1835 (2); ν(CC) 2117, ν(CO) 2005, 1865, 1830 (3); ν(CC) 2112, ν(CO) 2008, 1871, 1840 (4); ν(CC) 2116, ν(CO) 2002, 1854, 1826 (5). 15N, 31P, 183W and 195Pt NMR data are given in Table 3.
Preparation of [Rh(H)2(PPh3)}2(PTC)]
To a stirred solution of [Rh(H)(PPh3)4] (0.232 g, 0.182 mmol) in THF (5 ml) at room temperature was added a solution of 2-(4-pyridyl)thiazole-4-carboxylic acid (0.038 g, 0.184 mmol) in THF (3 ml). The mixture was then warmed to ~60 °C. After a few minutes at this temperature, a cream-colored precipitate began to form. After ~10 min, heating was discontinued, the mixture was allowed to cool and the product isolated and washed with diethyl ether to give a white powder. Yield 0,120 g (79 %). IR (compressed solid) ν(RhH) 2090, 2053, ν(CO) 1634; 1H NMR (CD3OD/THF (1:1) 300 K) 8.32 (mult, 2H, py 2,6), 7.67 (s, 1H, thiazole CH), 7.65 mult, 2H py 3,5), 7.47–7.26 (mults, 30H, PhP), −16.50 (mult, 1H, RhH), −19.78 (mult, 1H, RhH); 13C NMR (CD3OD/THF (1:1) 300 K) δ 168.09 (CO2), 165.57 (thiazole 2), 162.48 (thiazole 4), 150.38 (py 2,6), 139.77 (py 4), 134.80 (d (J(P,C) 6.6) PhP 2), 134.73 (d (J(P,C) 6.6) PhP 2′), 133.05 (d (J(P,C) 10.0) PhP 1), 131.17 (PhP 4), 129.84 (d (J(P,C) 13.2) PhP 1′), 129.39 (d (J(P,C) 4.9) PhP 3), 129.34 (d (J(P,C) 4.9) PhP 3′), 127.65 (thiazole 5), 123.45 py 3,5); 15N NMR (CD3OD/THF (1:1) 300 K) δ −107.11 (thiazole), −69.13 (py); 31P NMR (CD3OD/THF (1:1) 300 K) δ 42.97 (d (J(Rh,P) 119.7); 103Rh NMR (CD3OD/THF (1:1) 300 K) δ 509. Anal. Calc for C45H37N2O2P2RhS: C, 64.75; H, 4.47; N, 3.36. Found: C, 64.84; H, 4.54; N, 3.39.
Preparation of [Rh(H)2(PPh3)2(PTC)W(CO)4(P(4-MeC6H4)3)]·(MeOH) (6)
A solution of [Rh(H)2(PPh3)2(PTC)] (0.050 g, 0.052 mmol) and [W(CO)4(P(4-MeC6H4)3(CH3CN)] (0.038 g, 0.060 mmol) in THF/methanol (1:1, 6 ml) at room temperature was allowed to stand at room temperature for 5 days to give the product as a red powder/crystalline lumps. Yield 0.043 g (56 %), IR (compressed solid) ν(RhH) 2076, ν(CO) 2005, 1869, 1845, 1634; 1H NMR (CD3OD/THF (1:1) 300 K) δ 8.17 (mult, 2H, py 2,6), 7.71 (s, 1H, thiazole CH), 7.65 (mult, 2H, py 3,5), 7.45–7.08 (mults 42H ArP), 2.34 (s, 9H, Me), −16.54 (mult, 1H, RhH), −19.76 (mult, 1H, RhH); 15N NMR (CD3OD/THF (1:1) 300 K) δ −125.1 (py), −105.6 (thiazole); 31P NMR (CD3OD/THF (1:1) 300 K) δ 41.98 (d (J(Rh,P) 119)), 29.41 (s, W sat. (J(W,P) 39)); 103Rh NMR (CD3OD/THF (1:1) 300 K) 510; 183W NMR (CD3OD/THF (1:1) 300 K) δ 1463. Anal. Calc for C71H62N2O7P3RhSW: C, 58.13; H, 4.26; N, 1.91. Found: C, 58.88; H, 4.19; N, 1.96.
Results and discussion
Characterization of Pt–W complexes by NMR
None of the platinum-containing compounds could be obtained in a form suitable for structural characterization by X-ray diffraction; therefore, the following steps were taken using complex 1 to establish (by NMR) the major structural features illustrated in Scheme 1. (1) The binding of the acetylide to platinum was confirmed by the 13C spectrum, which shows spin coupling the dppp phosphorous atoms to both α and β acetylide carbons and coupling of 195Pt to the acetylide β carbon (2J(195Pt,13C) 300 Hz). (2) The binding of the pyridyl to tungsten was confirmed by 31P–15N-correlated HMQC in which the pyridyl nitrogen was observed by indirect detection using the phosphine attached to tungsten in complex 1. In a separate measurement, the same nitrogen was observed using pyridyl hydrogens for indirect detection, giving the same chemical shift for 15N (−143.6 ppm). (3) The 13C signal from the carbonyl positioned trans to PR3 on tungsten shows coupling to 31P with a magnitude 2J(31P,13C) trans = 33 Hz, while the carbonyls positioned cis to PR3 give 2J(31P,13C) cis of 4.1 and 7.0 Hz. In a complex having PR3 lying trans to pyridyl, all of the 31P–13C couplings will be cis. (4) The dppp 31P shows coupling to 195Pt and was used to obtain a 195Pt signal.

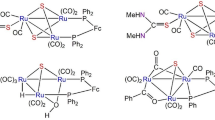
Chemical structure of platinum–tungsten complexes, PR3 = P(4-XC6H4)3 [X = H, (1), Me (2), OMe (3), F (4)], P(NMe2)3 (5)
Crystal structure of the Rh–W complex
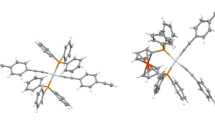
The chemical structure of [Rh(H)2(PPh3)2(μ-PTC)W(CO)4(P(4-MeC6H4)3)] (6) is shown in Scheme 2 and the crystal structure in Fig. 1. The crystal structure shows the coordination geometry at rhodium to be distorted octahedral with the hydrides, thiazole nitrogen and carboxylate oxygen occupying equatorial positions and the phosphines axial positions. The thiazolecarboxylate ‘bite’ angle, N–Rh–O, is 75.7° and the P–Rh–P angle is 162.2°, with the phosphines tilted away from the thiazolecarboxylate ligand. The pyridine ring is not coplanar with the thiazole (coplanarity might be expected to confer an advantage in terms of delocalization energy) but lies in a plane rotated by an angle of 35.50(12)° with respect to the plane of the thiazole. This alignment of the pyridyl group permits a π–π interaction [29] between the pyridyl ring and, on either side, offset so as partially to overlay the pyridyl ring, a phenyl group, one from a phosphine attached to rhodium, the other from the phosphine attached to tungsten (Fig. 2). The separation of the rings C65–C70 (phenyl of phosphine on rhodium) and N1, C5–C9 (pyridyl) is 3.960(3) (Å), indicating that atoms of the respective rings are almost in contact. Another π–π interaction is found between a phenyl group of the second phosphine attached to rhodium and the thiazole ring. The geometry at tungsten is slightly distorted octahedral, with the pyridyl and phosphine ligands lying cis to one another. Surprisingly, considering that the pyridyl and phosphine ligands are much bulkier than carbonyl, the N–W–P bond angle is significantly <90° (it is found to be 86.1°). A possible cause of this unexpected distortion might be the π–π interaction described above, which would tend to draw the pyridyl and phosphine ligands more closely together. The presence of the hydride ligands (on rhodium), for which evidence was found in the X-ray difference map, was confirmed by 1H NMR signals at δ −16.43 and −19.64 ppm, the former arising from the hydride positioned trans to nitrogen, the latter from the hydride positioned trans to oxygen.

Chemical structure of the rhodium–tungsten complex (6)

Perspective view of [(Ph3P)2Rh(H)2(μ-PTC)W(CO)4(P(4-MeC6H4)3)] (6). Thermal ellipsoids are shown at the 50 % probability level. Hydrogens and a co-crystallized methanol are omitted

Diagram of 6 showing π–π interactions between the pyridyl and phenyl groups of phosphines attached to the two metals
NMR study
In a complex having two metal atoms separated by a bridging ligand, the existence of an influence extending from one metal atom or its ligands to the metal atom (or its ligands) on the other side of the bridge should, in principle at least, be detectable by NMR spectroscopy if the two metals have isotopes with nuclear spin quantum number = 1/2, such as 183W and 195Pt. The influence might be expected to take the form of a contribution to the electronic shielding of one metal nucleus arising from the second metal and/or the ligands attached to it and might be transmitted through the bridge in the form of an inductive effect or arise from steric interactions between ligands attached to the respective metal atoms. The exchange of a ligand at one metal for a ligand having different electronic and/or steric properties should then induce measurable changes at the second metal. Complexes 1–5, having platinum and tungsten bridged by 4-pyridylacetylide, differ only in the identity of the phosphine attached to tungsten.
In terms of steric size, the phosphines attached to tungsten in complexes 1–4 (PR3 = P(4-XC6H4)3; X = H, Me, OMe, F) are experienced by the tungsten as sterically identical (Tolman cone angle [30] θ = 145°) but differ in their extension away from the tungsten and, hence, in their potential ability to interact through space with the phosphine attached to platinum. The phosphines PR3 in 1–4 vary to a moderate extent in their electron-donating ability, which is significantly lower than that of P(NMe2)3 in complex 5. Across the series 1–5, the platinum chemical shift shows little variation, with values of δ(195Pt) identical for 3, 4 and 5 (where there is a wide variation in the electronic properties of PR3) and only with PR3 = PPh3 is there a slight and barely significant response [a change of 6 ppm in δ(195Pt)] at platinum. This would appear to rule out an electronic interaction and suggest, if anything, a possible weak steric interaction. Variation in the coupling constant J(195Pt,31P) (2185 ± 5 Hz) falls within the limits of accuracy of the measurement.
At the tungsten atom, the chemical shift decreases by a significant amount (~60 ppm) on going from complexes containing triphenylphosphine and derivatives (1–4) to the complex of tris(dimethylamino)phosphine (5), consistent with the more strongly electron-donating properties of P(NMe2)3, and the tungsten–phosphorus spin-coupling constant increases by ~30 %. This increase in J(183W, 31P) arises from increased s character in the metal–phosphorus bond in cases where the phosphorus has nitrogen rather than carbon substituents. For complexes 1–4, there is a correlation between δ(183W) and J(183W,31P), where the sequence of increasing δ and increasing J, according to the substituent X in P(4-XC6H4)3, is Me < H < MeO < F. While this progression does not match the order of decreasing electron-donating ability given by the most commonly used measures of this property, namely the Tolman electronic parameter ν [30] and the Hammett parameter σp [31], (MeO > Me > H > F) the differences (in δ and in J) involved are small and may not be significant.
Complexes 2 and 6 have in common the fragment (4-C5H4N)W(CO)4(P(4-MeC6H4)3), which shows δ(183W) = 1,436.5 when attached to acetylide (in 2) and δ(183W) = 1,463 when bonded to thiazole (in 6). The much larger tungsten chemical shift difference found on exchanging the pyridyl substituents rather than the substituents on the phosphine phenyl have been documented elsewhere [32] but is unsurprising in view of the differing number of bonds separating the substituent and the metal.
Conclusion
A 195Pt NMR study of the complexes [(dppp)Pt{(μ-C2C5H4N)W(CO)4(PR3)}2], in which the phosphine PR3 is varied, provides no evidence for an electronic influence extending from the phosphine PR3 on tungsten through the bridge to platinum or for any form of steric interaction. The complex [(Ph3P)2Rh(H)2(μ-PTC)W(CO)4(P(4-MeC6H4)3)], characterized by X-ray crystallography, shows π–π interactions superimposed on the bridge connecting the two metal atoms. The interaction involves a phenyl group from a phosphine on rhodium, the bridging pyridyl and a tolyl group from the phosphine on tungsten and appears to give rise to a slight distortion in the tungsten coordination geometry.
Supplementary data
CCDC 913137 contains the crystallographic data for complex 6. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html, or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: (+44) 1223-336-033; or email: deposit@ccdc.cam.ac.uk.
References
Ziessel R, Hissler M, El-ghayoury A, Harriman A (1998) Coord Chem Rev 178–180:1251–1298
Long NJ, Williams CK (2003) Angew Chem Int Ed 42:2586–2617
Ceccon A, Santi S, Orian L, Bisello A (2004) Coord Chem Rev 248:683–724
Wu I-Y, Lin JT, Luo J, Sun S-S, Li C-S, Lin K-J, Tsai C, Hsu C-C, Lin J-L (1997) Organometallics 16:2038–2048
Kühn FE, Zuo J-L, Fabrizi de Biani F, Santos AM, Zhang Y, Zhao J, Sandulache A, Herdtweck E (2004) New J Chem 28:43–51
Ge Q, Hor TSA (2008) Dalton Trans 2929–2936
Ge Q, Dalton GT, Humphrey MG, Samoc M, Hor TSA (2009) Chem Asian J 4:998–1005
Ge Q, Corkery TC, Humphrey MG, Samoc M, Hor TSA (2009) Dalton Trans 6192–6200
Zuo J-L, Fabrizi de Biani F, Santos AM, Köhler K, Kühn FE (2003) Eur J Inorg Chem 449–452
Whiteford JA, Lu CV, Stang PJ (1997) J Am Chem Soc 119:2524–2533
Whiteford JA, Stang PJ, Huang SD (1998) Inorg Chem 37:5595–5601
Müller C, Whiteford JA, Stang PJ (1998) J Am Chem Soc 120:9827–9837
Packheiser R, Ecorchard P, Walfort B, Lang H (2008) J Organomet Chem 693:933–946
Grosshenny V, Harriman A, Ziessel R (1995) Angew Chem Int Ed 34:2705–2708
Grosshenny V, Harriman A, Hissler M, Ziessel R (1996) Plat Met Rev 40:26–35
Grosshenny V, Harriman A, Hissler M, Ziessel R (1996) Plat Met Rev 40:72–77
Harriman A, Ziessel R (1996) J Chem Soc Chem Commun 1707–1716
Chen Z-N, Fan Y, Ni J (2008) Dalton Trans 573–581
Tong J, Yu S-Y, Li H (2012) J Chem Soc Chem Commun 48:5343–5345
Halpin Y, Dini D, Younis Ahmed HM, Cassidy L, Browne WR, Vos JG (2010) Inorg Chem 49:2799–2807
Janka M, Anderson GK, Rath NP (2004) Organometallics 23:4382–4390
Adams RD, Perrin JL (1999) J Am Chem Soc 121:3984–3991
Ahmad N, Levison JJ, Robinson SD, Uttley MF (1974) Inorg Synth 15:45–64
Bax A, Griffey RH, Hawkins BL (1983) J Magn Reson 55:301–315
Carlton L, Emdin A, Lemmerer A, Fernandes MA (2008) Magn Reson Chem 46:S56–S62
APEX2, Version 2.0-1 (2005) Bruker AXS Inc., Madison
SAINT-NT, Version 6.0 (includes XPREP) (2005) Bruker AXS Inc., Madison, WI
SHELXTL, Version 5.1 (1999) Bruker AXS Inc., Madison, WI
Martinez CR, Iverson BL (2012) Chem Sci 3:2191–2201
Tolman CA (1977) Chem Rev 77:313–348
Hansch C, Leo A, Taft RW (1991) Chem Rev 91:165–195
Carlton L, Mokoena LV, Fernandes MA (2013) Magn Reson Chem (in press)
Acknowledgments
We thank the University of the Witwatersrand and the NRF for financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mampa, R.M., Fernandes, M.A. & Carlton, L. Preparation and NMR study of trimetallic complexes of platinum and tungsten bridged by 4-pyridylacetylide and a binuclear complex of rhodium and tungsten bridged by 2-(4-pyridyl)thiazole-4-carboxylate (PTC): structure of [(Ph3P)2Rh(H)2(μ-PTC)W(CO)4(P(4-MeC6H4)3)]. Transition Met Chem 38, 219–224 (2013). https://doi.org/10.1007/s11243-012-9681-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-012-9681-5