
Abstract
We performed detailed pharmacokinetic and pharmacodynamic modeling of REG1, an anticoagulation system composed of the direct factor IXa (FIXa) inhibitor pegnivacogin (RB006) and its matched active control agent anivamersen (RB007), with a focus on level of target inhibition to translate phase 1 results to phase 2 dose selection. We modeled early-phase clinical data relating weight-adjusted pegnivacogin dose and plasma concentration to prolongation of the activated partial thromboplastin time (aPTT). Using an in vitro calibration curve, percent FIXa inhibition was determined and related to aPTT prolongation and pegnivacogin dose and concentration. Similar methods were applied to relate anivamersen dose and level of reversal of pegnivacogin anticoagulation. Combined early-phase data suggested that ≥0.75 mg/kg pegnivacogin was associated with >99% inhibition of FIX activity and prolongation of plasma aPTT values ≈2.5 times above baseline, leading to selection of a 1 mg/kg dose for a phase 2a elective percutaneous coronary intervention study to achieve a high intensity of anticoagulation and minimize intersubject variability. Phase 2 validated our predictions, demonstrating 1 mg/kg pegnivacogin yielded plasma concentrations ≈25 μg/ml and >99% inhibition of FIX activity. The relationship between the anivamersen to pegnivacogin dose ratio and degree of pegnivacogin reversal was also validated. Our approach decreased the need for extensive dose–response studies, reducing the duration, complexity and cost of clinical development. The 1 mg/kg pegnivacogin dose and a range of anivamersen dose ratios are being tested in the phase 2b RADAR study (NCT00932100).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Selection of doses for phase 2 clinical studies is a critical juncture in the development of new therapeutics. Evaluation of inappropriate doses in initial phase 2 studies is a double-edged sword, with the potential to both jeopardize patient safety as well as misestimate the potential efficacy of the therapeutic if the selected doses are too high or too low. Selection of the appropriate dose range for antithrombotic therapies is especially important, as evaluation of doses below the therapeutic range can lead to thrombosis in treatment populations, whereas evaluation of doses beyond the therapeutic range can potentiate bleeding.
The REG1 Anticoagulant System (Regado Biosciences, Basking Ridge, NJ) is composed of the direct FIX/FIXa inhibitor pegnivacogin (RB006) and its matched, active control or reversal agent, anivamersen (RB007) [1]. Pegnivacogin is a modified ribonucleic acid (RNA) aptamer-based anticoagulant conjugated to a 40 kDa polyethylene glycol carrier that selectively and specifically targets factor IX/IXa (FIX/IXa), a coagulation protease required for thrombin generation [2]. Binding of pegnivacogin to FIX/IXa blocks association of FX with the FIXa/VIIIa complex, thereby inhibiting FX activation and eliciting an anticoagulant effect [3]. Pegnivacogin binds to FIX/IXa with a Kd of 3 nM, and equilibrium between free and bound pegnivacogin and FIX/IXa is established rapidly (within 5 min) after mixing. Onset of action and establishment of maximal plasma concentrations (Cmax) following bolus intravenous administration of pegnivacogin is within 2–5 min and its pharmacologic duration of action is >24 h at doses >0.75 mg/kg [1, 4, 5].
Anivamersen is a modified RNA oligonucleotide complementary to a portion of pegnivacogin which, upon binding to pegnivacogin, alters its conformation and rapidly neutralizes its pharmacodynamic effects [1]. Neutralization of pegnivacogin is established within 1–5 min following intravenous bolus administration of anivamersen, and is stable with no evidence of rebound anticoagulation over 14-day follow-up periods assessed in phase 1 clinical studies [1, 4, 5].
As a 2-component system, dose selection of the REG1 components for phase 2 studies is complicated by the potential for the interdependence of anivamersen dose on the administered pegnivacogin dose. Here we describe a translatable framework that combined analyses of a collection of clinical data derived across a series of phase I studies to develop models relating pegnivacogin and anivamersen dose, activated partial thromboplastin time (aPTT) prolongation, plasma drug concentrations, and inhibition of FIXa activity [1, 4, 5] to select a REG1 dosing strategy for phase 2 investigation. As phase 2 studies of REG1 targeted patients undergoing cardiac catheterization and percutaneous intervention (PCI), we sought to simplify phase 2 dose selection by identifying a single dose of pegnivacogin that would provide a consistently high level of intraprocedural anticoagulation within the target patient population, and a range of anivamersen doses that would enable evaluation of various anticoagulation reversal strategies post-procedure. The modeling approaches described here combined all phase I and 2a data to enable the successful efficient translation of appropriate doses for pegnivacogin and anivamersen into phase 2 studies, and to provide general guidance for selecting doses of pegnivacogin to achieve high-intensity anticoagulation through near complete inhibition of FIXa activity and anivamersen doses to predictably and reliably reverse this effect in a graded fashion.
Methods
The design, doses of drug administration, and timing of pharmacokinetic and pharmacodynamic sampling for the phase 1 program (Clin101 [1], Clin102 [4], and Clin103 [5]) and the phase 2a study (CLIN210 [6]) have been described in detail previously and are summarized in Table 1. In each study, drug (pegnivacogin or anivamersen) was consistently administered as a bolus intravenous injection over 1 min. Patients in all studies had dual intravenous access established prior to dosing and sample collection, with 1 site used exclusively for drug administration and the other for blood sampling. Pegnivacogin Cmax and pharmacodynamic effects are established within 5-min post-dose and remain essentially unchanged through at least 3 h post-dose. Therefore, 10- or 15-min post-dose samples were chosen for the present cross-study pharmacokinetic and pharmacokinetic/pharmacodynamic analysis because the data for these specific time points were available from each clinical study, and can be treated the same based upon the time to Cmax and stability of the plasma concentration over this time window.
Pharmacokinetic sample collection and bioanalytical methods
Samples for bioanalytical analysis were collected in K3EDTA tubes, mixed by inversion immediately upon withdrawal, and centrifuged within 15 min at 1500×g for 7 min at 4°C to generate plasma samples for analysis. Plasma was aliquoted into 1 ml aliquots, frozen at −70°C, and shipped on dry ice to a central laboratory for data collection.
Pegnivacogin plasma concentrations were measured using an ELISA method employing oligonucleotides complementary to pegnivacogin for capture and detection in human plasma as follows: a biotinylated deoxyribonucleic acid (DNA) capture probe complementary to the 3′-end of pegnivacogin (5′-[Biotin-TEG][spacer-18]GTGGAGGCAGCATTA-3′) and a digoxigenin labeled 2′O-methyl detection probe complementary to the 5′-end of pegnivacogin (5′-CGCGGUAUAGUCCAC[spacer-18][Digoxigenin]-3′) were added to plasma samples containing pegnivacogin and incubated at 37°C for approximately 2 h in a 96-well microtiter plate to allow hybridization of the probes to pegnivacogin. The mixture was then transferred to a neutravidin-coated microtiter plate to immobilize the pegnivacogin-probe complex on a surface. Detection was accomplished using an anti-digoxigenin antibody conjugated to alkaline phosphatase coupled with the fluorescent alkaline phosphatase substrate, AttoPhos (Promega Biosciences, Madison, WI). Fluorescence intensity was measured using a Spectramax Gemini fluorescence plate reader (Molecular Devices, Silicon Valley, CA), the signal of which was directly proportional to the amount of pegnivacogin present in the calibration standards, quality control samples, and study samples.
This assay was validated for specificity, working calibration range, dilution linearity, intra-batch precision, accuracy, freeze thaw stability, and long-term frozen storage. The validated linear range for the assay was 20–400 ng/ml pegnivacogin. Method performance during data collection, as determined by analysis of quality control samples included on each assay plate, was suitable. Typical within study assay performance characteristics include a percent coefficient of variation of 4.8–8.4% and a recovery of 89.7–98% over the concentration range of the quality control samples.
Pharmacokinetic analysis methods
Pegnivacogin Cmax values were determined by analysis of individual patient concentration time data. A power model was used to evaluate the linearity and dose proportionality of Cmax. This model assessed whether the slope of the relationship between the log of Cmax and the log of the dose was 1.0, as would be expected for a dose proportional exposure parameter. This demonstrated that Cmax was both dose proportional and linear over the range of doses studied based upon estimate of slope and 95% confidence intervals including a value of 1.0. Therefore, for the cross-study analysis, linear regression was used to determine the relationship between Cmax and weight-adjusted dose.
Pharmacodynamic sample collection and methods for coagulation measures
Plasma samples for coagulation measures were collected in 3.2% sodium citrate tubes, mixed immediately by gentle inversion, and centrifuged within 30 min of collection at 1000–1300×g for 15 min at room temperature to obtain plasma. Plasma was aliquoted into 1 ml aliquots, frozen at −70°C, and shipped on dry ice to a central laboratory for data collection for CLIN101 and 102. For CLIN103 and CLIN210, samples were not frozen and were assayed within 3 h of collection at the clinical core lab at the enrolling site.
For CLIN101 and CLIN102, aPTT assays were performed on a Stago STA analyzer (Stago, Parsippany, NJ) using STA-PTT A reagents at a central laboratory (Icon Laboratories, Farmingdale, NY). FIX activity assays for CLIN101 and CLIN102 were collected on the same instrumentation using the STA-PTT A reagent and the STA FIX Deficient Substrate as the plasma depleted in FIX to generate the FIX activity reference curve. For CLIN103, aPTT assays were performed at the enrolling clinical research unit (PPD, Austin, TX) on a Sysmex CA1500 using Actin FSL aPTT reagent (Dade-Behring, Newark, DE). CLIN210 was a multicenter study in which aPTT assays were performed locally at the enrolling site. Subsequently, specific information regarding instrumentation and reagents for plasma aPTT assays was not collected.
Samples for whole blood coagulation assays were collected fresh and tested immediately. Whole blood point-of-care (POC) aPTT and activated clotting time (ACT) were performed on a Hemochron Signature Plus Microcoagulation System (International Technidyne Corporation, Parsippany, NJ). POC aPTTs were performed using the Hemochron Jr. APTT test cuvette per the manufacturer’s instructions, and data are presented in seconds. ACTs were performed using the Hemochron Jr. ACT-LR test cuvette per the manufacturer’s instructions. Hemochron Signature Plus Microcoagulation Systems were purchased by Regado Biosciences and provided to each enrolling site in CLIN210 for use in collection of whole blood coagulation measures during this study.
Pharmacokinetic and pharmacodynamic analysis methods
Pharmacokinetic/pharmacodynamic analysis was conducted using WinNonlin version 5.2 (Pharsight Corporation, Mountain View, CA). For analysis of the relationship between aPTT and the plasma concentration of pegnivacogin, the sigmoidal Emax model, E = E0 + (Emax − E0)[Cγ/(Cγ+EC γ50 )] was employed, where concentration (C) is in μg/ml. In this analysis, the baseline (E0) is the aPTT ratio at time zero which is equal to 1, the Emax is the estimated maximum aPTT ratio, the ED50 is the pegnivacogin dose providing 50% of the maximum effect, and gamma (γ) represents the slope of the dose–response curve.
To determine the relationship between percent inhibition of FIXa activity and pegnivacogin dose and concentration, an indirect calculation approach was used, as direct assays of FIXa activity are not available. Plasma FIX assay calibration curves generated at the clinical core laboratory (Icon Laboratories, Farmingdale, NY) during the sample analysis in the CLIN101 and CLIN102 studies were obtained, and the mean of the relative aPTT prolongation versus FIX level were plotted. The mean calibration curves were fit per the FIX assay kit, which yielded the following relationship between percent of FIX Activity and relative aPTT Log(c)= (−5.618 * Log(t)) + 12.28, where c is the percent of FIX activity and t is the relative aPTT value. The percent of FIX activity for individual plasma samples isolated from pegnivacogin-treated subjects was then imputed from the calibration curve based on the relative aPTT prolongation of each sample, and the percent of inhibition of FIX calculated (e.g., 100-percent FIX activity = percent of FIX inhibition).
Results
The CLIN101 [1], 102 [4], and 103 [5] studies individually demonstrated a dose relationship between the amount of pegnivacogin administered and the prolongation of aPTT. However, these analyses did not define the relationships between pegnivacogin and anivamersen dose, concentration, and level of target inhibition necessary to translate the independent phase 1 observations into appropriate models for dose selection of both components of REG1 for phase 2 studies. Therefore, we undertook this cross-study pharmacokinetic and pharmacokinetic/pharmacodynamic analysis to develop the necessary models to establish these important relationships.
Key characteristics of the clinical studies from which data were collected for this cross-study analysis, including design, treatment, numbers of subjects, and sampling time periods and methodologies are listed in Table 1.
Assessment of pegnivacogin dosing in CLIN101 and CLIN102
Based upon the structure of pegnivacogin, we expected its distribution to be predominantly within the vascular compartment, and therefore anticipated a predictable, linear relationship between the weight-adjusted pegnivacogin dose and plasma concentration. Accordingly, non-weight-based pegnivacogin dosing was utilized in the CLIN101 and 102 studies to provide a broad range of weight-adjusted doses for cross-study pharmacokinetic and pharmacodynamic analyses. Analysis of Cmax pegnivacogin versus weight-adjusted pegnivacogin dose in CLIN101 and 102 is shown in Fig. 1a, demonstrating a linear relationship between weight-adjusted pegnivacogin dose and Cmax, and predicting pegnivacogin Cmax (in μg/ml) = 27.98 (pegnivacogin dose mg/kg) + 0.217.

Cross-study pharmacokinetic, and pharmacokinetic/pharmacodynamic assessments from CLIN101 and CLIN102. a Relationship between maximal pegnivacogin concentration achieved (Cmax) and weight-adjusted pegnivacogin dose. b Relationship between relative increase in aPTT and pegnivacogin concentration. Open diamonds represent data from CLIN101 and closed squares represent data from CLIN102
Having established the relationship between the weight-adjusted pegnivacogin dose and plasma concentration achieved, we determined the relationship between pegnivacogin concentration and aPTT prolongation. Pegnivacogin concentrations were plotted versus relative aPTT (Fig. 1b) and analyzed using a sigmoidal Emax model. We calculated an EC50 of 14 μg/ml with a maximal predicted effect of 2.42-fold increase in aPTT above baseline. While informative, the precision of this analysis is limited by the lack of a fully defined upper plateau. Additionally, since pegnivacogin is the first selective direct FIX/IXa inhibitor to enter clinical development, interpretation of the resultant model parameters in relation to dose selection is not possible in the absence of defining the relationship of the aPTT prolongation by pegnivacogin to the level of FIXa inhibition.
Relationship between FIX activity and aPTT
To define the relationship between pegnivacogin dose and level of FIX/IXa inhibition, we first developed a model relating the level of FIX activity to prolongation of the aPTT. The CLIN101 and 102 studies used the same clinical core laboratory with the same aPTT reagent and methodology for the plasma FIX assay and aPTT measures from subjects’ samples. Therefore, we were able to utilize the calibration curves from several FIX assay runs performed during the CLIN101 and 102 studies to define the relationship between the relative aPTT prolongation and level of plasma FIXa activity. The mean fit of the log-linear calibration curves derived from FIX assays performed during data capture in the CLIN101 and 102 studies is depicted graphically in Fig. 2. This calibration curve shows that, using the aPTT assay implemented across CLIN101 and CLIN102, depletion of FIX activity to <1% normal (>99% inhibition) is associated with a 2.5-fold prolongation of the aPTT, a value consistent with the observed maximum effect of pegnivacogin in the dose response plot (Fig. 1b). The level of pegnivacogin mediated FIX/IXa inhibition achieved in CLIN101 and CLIN102 can then be calculated by using the observed relative increase in aPTT to impute the FIX activity remaining in these samples, followed by subtraction of this value from 100%.

Relationship between percent of FIX activity and the relative increase in aPTT. Mean FIX assay calibration curves obtained during data collection for CLIN101 and CLIN102
The relationship between the percent of FIX/IXa inhibition and pegnivacogin dose and concentration are shown Fig. 3a and b, respectively. Based on these models, pegnivacogin doses of ≥0.75 mg/kg (Fig. 3a) and pegnivacogin plasma concentrations ≥15 μg/ml (Fig. 3b) are predicted to lead to essentially complete inhibition of plasma FIXa activity. The observed difference between the pegnivacogin plasma concentration at which maximal FIXa inhibition is established and the predicted Cmax for the dose at which this level of inhibition is achieved is due to the non-linear relationship between pegnivacogin concentration and aPTT prolongation as the maximum effect is approached.

Cross-study modeling of relationship between level of FIXa inhibition and pegnivacogin dose and concentration. a Percent of FIXa inhibition versus weight-adjusted pegnivacogin dose. b Percent of FIXa inhibition versus pegnivacogin plasma concentration. Samples in this analysis were collected 15 min post-dose for CLIN101 and 10 min post-dose for CLIN102. Full data sets for these draw times were available for all pegnivacogin-treated subjects at this timepoint in the respective studies, and pegnivacogin concentrations at both sampling times are representative of Cmax. Open diamonds represent data from CLIN101 and closed squares represent data from CLIN102
These findings were used to transition to a weight-based dose of 0.75 mg/kg pegnivacogin in the CLIN103 study. Median (25th, 75th percentiles) (21.67; [17.14, 23.63 μg/ml]) and mean (21.66; 95% confidence interval [CI] 19.51, 23.81 μg/ml) pegnivacogin concentrations for the first day of pegnivacogin dosing (n = 29) were in excellent agreement with the predicted Cmax of 21.2 μg/ml, yielding an average increase in the aPTT of 241 ± 22% (approximately 2.5-fold increase over baseline) [5]. No bleeding or other safety signals were related to this dose of pegnivacogin and the level of FIXa inhibition observed in the CLIN103 study.
Relationship between anivamersen dosing and pegnivacogin reversal
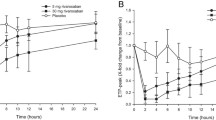
To assess anticoagulant reversal using anivamersen, subjects participating in CLIN101 and CLIN102 received a 2:1 weight:weight dose ratio of anivamersen:pegnivacogin, corresponding to a molar ratio of approximately 20:1. This anivamersen dose resulted in complete reversal of the pharmacodynamic effect of pegnivacogin within 1–5 min of anivamersen administration across a range of pegnivacogin doses, an effect that remained durable throughout the 7-day follow-up period [1, 4]. The observation that a fixed weight:weight anivamersen:pegnivacogin dose ratio was effective across a range of pegnivacogin doses confirmed prior in vitro observations, simplifying anivamersen dose selection. To test this observation and extend our understanding of the anivamersen dose response, a range of anivamersen doses were evaluated against a fixed weight-adjusted dose of pegnivacogin in the CLIN103 trial [5]. Anivamersen administration at dose ratios of anivamersen:pegnivacogin of 2:1 and 1:1 fully and rapidly reversed the observed pharmacodynamic effects, returning aPTT values to baseline values within 1–5 min. Complete pegnivacogin reversal by anivamersen remained durable throughout the 14-day follow-up period. Lower doses (weight-based ratios) of anivamersen produced a graded reversal response, with ratios of 0.5:1, 0.3:1, 0.2:1, and 0.125:1 reversing 84 ± 2.7, 74 ± 5.9, 51 ± 6.4, and 41 ± 8.4% of the observed maximum increase in aPTT from baseline, respectively, (Fig. 4) [5] confirming the utility of the weight:weight dose ratio approach for anivamersen. Additionally, following partial reversal of pegnivacogin by anivamersen, the remaining anticoagulant activity decayed in a manner consistent with the pharmacodynamics observed following administration of pegnivacogin alone.

Dose response of anivamersen administration on pegnivacogin-induced prolongation of aPTT. Anivamersen doses are presented as relative mg dose:dose ratio of pegnivacogin dose administered in CLIN103. Pegnivacogin dose employed in CLIN103 was 0.75 mg/kg. Ani anivamersen, Peg pegnivacogin
CLIN210: First-In-Man PCI Study
High intensity anticoagulation is required for PCI; however, high levels of target inhibition are rarely tested due to concerns related to increasing bleeding risk. Given the availability of rapid and effective anivamersen-mediated pegnivacogin reversal, we selected a dosing strategy that targeted near complete FIXa inhibition during PCI followed by anivamersen administration shortly after procedure completion to mitigate bleeding risk. Based on the previously presented analysis, a pegnivacogin dose of 0.75 mg/kg was predicted to approximate a threshold at which complete FIXa inhibition could be reliably achieved. To minimize intersubject variability in the anticoagulation level achieved in a patient population and given the availability of reversal, a modestly higher dose of 1 mg/kg pegnivacogin was chosen for the CLIN210 (NCT00715455) study [6].
CLIN210 was the first clinical use of pegnivacogin as the sole anticoagulant during PCI. For anivamersen dosing in CLIN210, a total dose of 2 mg/kg (2:1 weight:weight pegnivacogin dose ratio) was conservatively chosen to effect full pegnivacogin reversal following PCI. In the first cohort (n = 10), this dose was administered in 2 parts: the first dose (0.2 mg/kg), targeting 50% reversal of the aPTT was given immediately after PCI, with the remaining 1.8 mg/kg anivamersen administered 4 h post-PCI to fully reverse pegnivacogin activity. A second cohort (n = 10) was administered a 100% reversal dose (2 mg/kg) immediately post-procedure.
Median (25th, 75th) (26.15 [8.65, 30.38 μg/ml]) and mean (24.63 [95% CI 21.49, 27.76] μg/ml) pegnivacogin concentrations at the beginning of the procedure were in excellent agreement with the predicted Cmax of 28.2 μg/ml. Peak levels were achieved within 5 min of drug administration (Fig. 5a) and did not significantly differ from those observed at the end of PCI (median concentration 26.05 [25th, 75th 19.03, 28.23] μg/ml). Administration of anivamersen resulted in the expected dose-dependent elimination of plasma pegnivacogin (Fig. 5a). The partial reversal 0.2:1 dose ratio of anivamersen reduced the mean measurable free pegnivacogin concentration to 8.7 ± 2.3 μg/ml 15 min post-dosing. Full reversal doses of anivamersen reduced free pegnivacogin to <20 ng/ml, the limit of quantification of the assay.

Pharmacokinetic and pharmacodynamic assessments in elective PCI patients in CLIN210. a Pegnivacogin plasma concentrations following 1 mg/kg dose during PCI, and either partial then full-reversing doses or a single full reversing dose of anivamersen post-PCI. b Relative increase in plasma aPTT during PCI following 1 mg/kg pegnivacogin dose, and either partial then full-reversing doses or a single full reversing dose of anivamersen post-PCI. c Prolongation of whole blood aPTT during PCI following 1 mg/kg pegnivacogin dose, and either partial then full-reversing doses or a single full reversing dose of anivamersen post-PCI. d ACT measures following 1 mg/kg dose of pegnivacogin during PCI, and either partial then full-reversing or a single full reversing dose of anivamersen post-PCI. Open squares represent subjects assigned to the group receiving sequential doses of 0.2 mg:mg followed by 1.8 mg:mg anivamersen:pegnivacogin; closed circles represent subjects assigned to the group receiving a single, full reversing dose of 2 mg:mg pegnivacogin:anivamersen. Data are presented as median ± 25th, 75th percentiles
Consistent with the plasma concentration data, there was a rapid prolongation of the plasma aPTT, reaching a maximal value of 2.5 times the baseline within 5 min after pegnivacogin administration (Fig. 5b) and remaining essentially unchanged throughout the PCI procedure, validating the dose chosen based on modeling of the phase I experience presented in Figs. 1–4. Administration of a 0.2:1 dose ratio of anivamersen resulted in a stepwise change in pharmacodynamic effect (Fig. 5b), resulting in mean reversal of 51 ± 14% of the aPTT prolongation. This result confirmed the anivamersen dose–response observed in the CLIN103 study and that weight-based anivamersen:pegnivacogin dosing reliably predicts reversal, even with the change in pegnivacogin dose between CLIN103 and CLIN210. Rapid and complete reversal of pegnivacogin was achieved using either a 2:1 dose ratio of anivamersen or sequential 0.2:1 and 1.8:1 doses. Whole blood POC aPTT (Fig. 5c) and ACT measures (Fig. 5d) corroborate previous findings. Coagulation measures remained consistent 15 min post-pegnivacogin administration and at the end of the PCI procedure, demonstrating the feasibility of using a single dose of pegnivacogin to achieve and maintain adequate levels of anticoagulation during PCI. Furthermore, as observed with the plasma aPTT assay, both whole blood coagulation measures were reduced in a dose-dependent manner following anivamersen administration. All pharmacodynamic measures were consistent with pairwise pegnivacogin plasma concentration measures and modeling of pharmacokinetic/pharmacodynamic data from phase 1 studies.
Discussion
The safety and effectiveness of antithrombotic therapies are critically dependent on proper dose selection. Therefore, a thorough characterization and understanding of the relationship between plasma concentrations, measurable pharmacodynamic effects, and levels of target inhibition is crucial for the translation of doses of such drugs from phase 1 to phase 2 to ensure proper evaluation of efficacy parameters while protecting the safety of subjects.
The REG1 system is novel in its target of action (FIXa), the aptameric formulation of its anticoagulant component pegnivacogin, and the availability of a specific reversal agent, anivamersen. Here we describe the steps taken to establish the relationship between pegnivacogin dose, the effect on established measures of coagulation, the achieved plasma drug concentrations, and the degree of target inhibition, as well as a simplified dosing strategy and effective dose ranges for anivamersen which enabled rapid progression from early phase studies to dose selection for later phase trials.
Modeling in support of pegnivacogin dose selection
Loss of FIX activity, as observed in patients with hemophilia B, leads to prolongation of the aPTT. Initial phase 1 studies of REG1 demonstrated a clear dose-dependent prolongation of the aPTT by pegnivacogin, and rapid reversal of this pharmacodynamic effect upon anivamersen dosing [1, 4, 5]. To synthesize the pharmacokinetic and pharmacodynamic data generated in these 3 phase 1 studies of REG1 into a coherent model relating dose, plasma concentration and degree of target inhibition, we first focused on defining the relationship between pegnivacogin dose, plasma concentration, and aPTT prolongation by performing a cross-study analysis of data obtained in the CLIN101 and 102 studies (Fig. 1). This analysis established a linear relationship between weight-adjusted dose of pegnivacogin and its plasma concentration, and established the concentration response relationship of pegnivacogin in the aPTT.
To establish the relationship between these parameters and degree of FIX/IXa inhibition, we defined the relationship between aPTT prolongation and the amount of FIX activity present using the FIX assay calibration curves collected by the central clinical core laboratory during the conduct of CLIN101 and CLIN102. The FIX assay calibration curves demonstrated that progressive FIX depletion caused aPTT prolongation in a log-linear fashion, approaching a maximal effect at approximately 2.5 times the normal value, which correlates with >99% inhibition of FIX activity (<1% residual activity) (Fig. 2). Since all the plasma coagulation analyses for CLIN101 and 102 were performed at the same site using the same methodology and reagents, we were able to use this calibration curve to impute the level of pegnivacogin-induced FIX/IXa inhibition based on the observed aPTT prolongation in individual subject samples. This analysis established a direct relationship between pegnivacogin dose, concentration and degree of FIX/IXa inhibition (Fig. 3), providing a clear relationship from which the dose and target plasma concentration required to achieve the desired degree of FIX/IXa inhibition could be selected.
To verify key outputs of the above modeling, including the relationship between weight-adjusted pegnivacogin dose, Cmax and aPTT prolongation, CLIN103 utilized a weight-based dose of pegnivacogin of 0.75 mg/kg [5] which, based on the modeling presented in Fig. 3, was predicted to represent the minimal dose at which essentially complete FIXa inhibition is achieved. Analysis of pharmacokinetic and pharmacodynamic parameters in CLIN103 verified the accuracy of the Cmax model and the pharmacokinetic/pharmacodynamic models that predicted this dose would provide an aPTT prolongation consistent with complete inhibition of FIX/IXa activity.
Pegnivacogin dose selection for Phase 2 in PCI
Selection of pegnivacogin dosing for an initial phase 2a study in elective PCI, as well as for potential follow-on studies in acute coronary syndromes (ACS), was based on the desire to achieve a high level of intraprocedural antithrombotic therapy to prevent ischemic complications and halt ongoing thrombus formation, as well as the desire to limit interpatient variability. The decision to target near complete FIX/IXa inhibition was facilitated by the availability of immediate anivamersen-mediated pegnivacogin reversal, should bleeding arise or the need for high level target inhibition change. In addition, given that pegnivacogin is the first FIX/IXa inhibitor in advanced clinical study, we anticipated that the achievement of maximal levels of target (FIXa) inhibition would simplify interpretation of clinical results and validation of FIX/IXa as a target for antithrombotic therapy.
The pegnivacogin dose selected (1 mg/kg) was modestly above the minimal predicted dose required for complete FIXa inhibition and resulted in mean pegnivacogin concentrations of 26.1 μg/ml, in excellent agreement with the expected values. The resultant pharmacodynamic effects (mean increase in aPTT to 2.5× normal) were consistent with models demonstrating that plasma pegnivacogin concentrations ≥15 μg/ml are associated with maximal inhibition of FIX activity and maximal aPTT prolongation [6]. No untoward ischemic events, including stent thrombosis, were observed during this study, demonstrating the feasibility of using pegnivacogin as an anticoagulant to support PCI.
Modeling in support of anivamersen reversal dose selection
Translation of the dosing of a 2-component system such as REG1 from phase 1 to phase 2 studies is potentially complicated by the interdependence of the doses of the 2 agents. The 1:1 binding mechanism of anivamersen to pegnivacogin suggested we could simplify this potential hurdle by using weight:weight dose ratios of anivamersen:pegnivacogin, as the fraction of pegnivacogin bound by anivamersen should remain consistent across a wide range of pegnivacogin doses. Initial in vitro experiments demonstrated that full reversal of pegnivacogin-mediated FIXa inhibition was observed at a 0.5:1 weight:weight anivamersen:pegnivacogin ratio, corresponding to a molar ratio of approximately 5:1. As anivamersen is rapidly metabolized (half-life ≈5 min) and has no known biological effects, a 4-fold excess above the projected minimum anivamersen dose required for complete reversal was targeted for initial clinical studies. The resulting weight-based regimen of 2:1 anivamersen:pegnivacogin achieved the predicted complete reversal of anticoagulation parameters across a wide range of weight-adjusted pegnivacogin doses in the CLIN101 and CLIN102 studies [1, 4], providing initial confirmation of this approach to anivamersen dosing.
To provide additional confirmation of this approach and extend our understanding of the clinical dose relationship between anivamersen dose and pegnivacogin reversal, CLIN103 tested a wide range of anivamersen doses against a fixed, weight-adjusted dose of pegnivacogin (Fig. 4). This study confirmed the dose ratio concept by demonstrating full reversal of pegnivacogin at 2:1 and 1:1 dose ratios of anivamersen, with a stepwise functional decrease in the degree of pegnivacogin reversal as dose ratios were reduced below 1:1 down to 0.125:1 [5].
Anivamersen dose selection for phase 2 in PCI
In selecting the anivamersen dose in CLIN210, consideration was given to the clinical safety concerns associated with both immediate reversal of anticoagulation post-PCI balanced against the need for adequate normalization of coagulation parameters to allow safe femoral sheath removal. Therefore, a 2:1 weight:weight dose ratio of anivamersen:pegnivacogin was selected to provide complete pegnivacogin reversal prior to sheath removal, and 2 anivamersen dosing strategies were explored sequentially in CLIN210 to evaluate the tolerability of complete reversal post-PCI. In the first cohort, a stepwise reversal approach was evaluated in which an anivamersen dose ratio of 0.2:1 targeting 50% reversal was first administered immediately after PCI, followed 4 h later by administration of the additional 1.8:1 dose ratio to provide full normalization of coagulation parameters. In the second cohort, the 2:1 anivamersen dose ratio was administered immediately after completion of PCI to provide complete reversal. Pharmacokinetic and pharmacodynamic measures following anivamersen dosing in both cohorts of CLIN210 confirmed the translatability of the dose ratio approach and selected doses of anivamersen (Fig. 5). The initial partial reversing dose of anivamersen resulted in a consistent fractional decrease in pegnivacogin concentration, and full reversal doses resulted in elimination of free plasma pegnivacogin. Likewise, the initial 0.2:1 dose provided the targeted 50% reversal in aPTT prolongation, and the 2:1 dose administered in parts or as a single bolus provided complete reversal of pegnivacogin anticoagulation.
Limitations
The selected aPTT-based modeling approach was successful for dose selection for elective PCI and ACS indications, where the high intensity anticoagulation desired would result in a significant increase in the aPTT. However, because prolongation of the aPTT requires a significant diminution of FIX activity, the current approach may not translate well to other indications for which lower degrees of FIXa inhibition may be sufficient and desired. For these indications, use of pharmacodynamic read-outs responsive to lower levels of FIXa inhibition would be preferable to better determine an optimal dose. Additionally, the response curve described in Fig. 2 will be specific for a particular reagent and aPTT assay. While similar results might be expected across aPTT reagents, inferring an intermediate degree of FIXa inhibition for any given assay would require re-establishment of the FIX activity–aPTT relationship, limiting the utility of non-standardized aPTT assays to reliably predict degree of FIXa inhibition. Maximal degrees of FIXa inhibition can be inferred from an aPTT if performance of the specific reagent in FIX-depleted plasma is provided as part of the package insert. Nonetheless, the consistent results across a variety of studies and assays in CLIN101/102, CLIN103, and CLIN210 suggest that increases of aPTT >2× over baseline is reflective of and could be used as a reliable measure of near complete FIX inhibition.
Conclusions
We combined pharmacokinetic and pharmacodynamic data obtained in a series of phase 1 studies to model the dose-anticoagulant response for pegnivacogin, a direct FIXa inhibitor, establishing a relationship between its dose, concentration, effect on aPTT, and the degree of target (FIXa) inhibition. In parallel, data from these studies were also modeled to evaluate a weight:weight dose ratio approach for anivamersen, the specific active control agent for pegnivacogin, and to determine the relationship between the anivamersen:pegnivacogin dose ratio and degree of reversal of pegnivacogin anticoagulation. Based on cumulative data from 3 phase 1 studies, a pegnivacogin dose of 1 mg/kg was selected for a phase 2a trial of elective PCI, which in turn validated the relationship between pegnivacogin dose, concentration, inhibition of FIX activity, and prolongation of aPTT-based measures of coagulation [7]. Likewise, a weight:weight dose ratio approach for anivamersen dosing relative to administered pegnivacogin was confirmed in these 3 phase 1 studies, simplifying the selection of appropriate reversal doses for phase 2 to achieve targeted partial or complete pegnivacogin reversal levels. This dosing strategy and dose levels were validated in the phase 2a study in parallel with the 1 mg/kg pegnivacogin dose. The information obtained in early phase development has provided a platform for RADAR (NCT00932100), a larger phase 2b study of REG1 in patients with ACS [8]. Our modeling strategy also provided information sufficient for pegnivacogin dose selection without the requirement for pegnivacogin dose–response studies in an ACS patient population, greatly decreasing the overall cost and time to develop REG1 for an ACS indication.
Addendum
Drs. Povsic, Rusconi, and Becker were responsible for writing of this manuscript. Drs. Cohen, Chan, Alexander, Harrington and Becker were responsible for execution and coordination of the clinical trials in the manuscript. Drs. Zelenkofske, Becker, Rusconi, and Wargin were responsible for data analysis.
References
Dyke CK, Steinhubl SR, Kleiman NS, Cannon RO, Aberle LG, Lin M, Myles SK, Melloni C, Harrington RA, Alexander JH, Becker RC, Rusconi CP (2006) First-in-human experience of an antidote-controlled anticoagulant using RNA aptamer technology: a phase 1a pharmacodynamic evaluation of a drug-antidote pair for the controlled regulation of factor IXa activity. Circulation 114(23):2490–2497
Howard EL, Becker KCD, Rusconi CP, Becker RC (2007) Factor IXa inhibitors as novel anticoagulants. Arterioscler Thromb Vasc Biol 27(4):722–727
Rusconi CP, Scardino E, Layzer J, Pitoc GA, Ortel TL, Monroe D, Sullenger BA (2002) RNA aptamers as reversible antagonists of coagulation factor IXa. Nature 419(6902):90–94
Chan MY, Cohen MG, Dyke CK, Myles SK, Aberle LG, Lin M, Walder J, Steinhubl SR, Gilchrist IC, Kleiman NS, Vorchheimer DA, Chronos N, Melloni C, Alexander JH, Harrington RA, Tonkens RM, Becker RC, Rusconi CP (2008) Phase 1b randomized study of antidote-controlled modulation of factor IXa activity in patients with stable coronary artery disease. Circulation 117(22):2865–2874
Chan MY, Rusconi CP, Alexander JH, Tonkens RM, Harrington RA, Becker RC (2008) A randomized, repeat-dose, pharmacodynamic and safety study of an antidote-controlled factor IXa inhibitor. J Thromb Haemost 6(5):789–796
Cohen MG, Purdy DA, Rossi JS, Grinfeld LR, Myles SK, Aberle LG, Greenbaum AB, Fry E, Chan MY, Tonkens RM, Zelenkofske S, Alexander JH, Harrington RA, Rusconi CP, Becker RC (2010) First clinical application of an actively reversible direct factor IXa inhibitor as an anticoagulation strategy in patients undergoing percutaneous intervention. Circulation 122(6):614–622
Gopinath SCB, Shikamoto Y, Mizuno H, Kumar PKR (2006) A potent anti-coagulant RNA aptamer inhibits blood coagulation by specficially blocking the extrinsic clotting pathway. Thromb Haemost 95(5):767–771
Povsic TJ, Cohen MG, Mehran R, Buller CE, Bode C, Cornel JH, Kasprzak JD, Montalescot G, Joseph D, Wargin WA, Rusconi CP, Zelenkofske SL, Becker RC, Alexander JH (2011) A randomized, partially blinded, multicenter, active-controlled, dose-ranging study assessing the safety, efficacy, and pharmacodynamics of the REG1 anticoagulation system in patients with acute coronary syndromes: design and rationale of the RADAR phase IIb trial. Am Heart J 161(2):261–268
Acknowledgments
We would like to thank the CLIN101, 102, 103, and 210 clinical site investigators and research team personnel who diligently executed the study protocols, provided important feedback for future study design, and produced high quality data to support the continued development of the REG1 System. We also thank Elizabeth Cook for exceptional editorial input and assistance.
Disclosure
Dr. Rusconi is a founder, employee, and stockholder in REGADO Biosciences. Dr. Zelenkofske is an employee and stockholder in REGADO Biosciences. Dr. Wargin is a consultant for REGADO Biosciences. Drs. Becker, Povsic, Harrington, and Alexander are employed by the Duke Clinical Research Institute which has received research funding from REGADO Biosciences.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Povsic, T.J., Cohen, M.G., Chan, M.Y. et al. Dose Selection for a Direct and Selective Factor IXa Inhibitor and its Complementary Reversal Agent: Translating Pharmacokinetic and Pharmacodynamic Properties of the REG1 System to Clinical Trial Design. J Thromb Thrombolysis 32, 21–31 (2011). https://doi.org/10.1007/s11239-011-0588-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11239-011-0588-3