
Abstract
Four acridinium/acridine derivatives have been prepared by mechanochemical synthesis and their crystal structure, supramolecular framework, interaction energy calculation, thermal analysis, and photophysical properties are studied and presented in this manuscript. In the crystal structure, the one- and two-dimensional supramolecular framework is constructed via various strong and weak intermolecular interactions such as N‒H…Cl, O‒H…N, O‒H…Cl, C‒H…Cl, C‒H…O, C‒H…π, and π…π, respectively. The contribution of intermolecular interaction in the three-dimensional molecular packing is studied by the Hirshfeld surface analysis. The calculated total energy value of intermolecular interaction/contacts observed between the molecular pairs in the acridine compound is stronger than the energy value of intermolecular interaction/contacts of acridinium derivatives. The energy value of π…π contacts exhibited between the molecular pairs is significantly stronger than other weak interactions. The thermogravimetric analysis reveals that the acridinium derivatives degrade in three steps whereas the acridine compound undergoes a single-step degradation. Studies show that the acridinium derivatives exhibit a better photoluminescence quantum yield when compared to the acridine compound, and the acridine compound experiences a photoluminescence quenching due to charge transfer interactions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Acridines are a class of heterocyclic compounds with three fused six-membered rings exhibiting a planar aromatic surface. Acridines are structurally analogous to anthracene with a N atom replacing the C atom in the 10th position. Acridine compound evolved as the byproduct of aniline dye manufacture and had been used in clinical medicine in the late nineteenth century. Over the years, acridine derivatives have been known to exhibit a series of bioactivities such as antimicrobial, anticancer, antimalarial, antidepressant, antifungal, and antibacterial [1,2,3,4,5,6]. Recent research on acridine/acridinium derivatives has been found mainly on their use as anticancer drugs, due to the planar ring structure that grants the ability to diffuse through the cell membrane and intercalate with the DNA [7]. It is reported that the photocytotoxicity of propylacridine acts against leukemia with 9-(2′-hydroxyethylamino)-4-methyl-1-nitroacridine acts as a promising antitumor drug against UDP-glucuronosyltransferases (UGTs) [8]. Interestingly, acridine derivatives are capable of displaying chemiluminescence properties, and acridinium cations with substitutions at the N10 atom and electron-attracting substituents at the C9 atom are capable of exhibiting good light emission or chemiluminescence property [9, 10]. The efficiency towards the emission of light or chemiluminescence is greatly influenced by the nature of various substitutions such as nucleophilic species, oxidizing agents, and other entities at the C9 atom [11, 12]. Acridinium derivatives with N-alkyl substituents are widely used in medical and biological analyses due to their ability to react with appropriate macromolecular fragments [13]. The chemiluminescence properties of cation or acridinium derivatives are comparably efficient to neutral acridine derivatives. Apart from medical importance, these types of acridine luminescence-efficient materials have received attention in the past few years due to their intriguing applications in optoelectronic devices and biosensors [13,14,15]. Intermolecular hydrogen bonds such as N‒H…O, O‒H…O, N‒H…X (X = Cl, Br, I, S), halogen bonds, and other weak interactions such as C‒H…π and π…π contacts are of fundamental importance in self-assembling the molecules in a crystalline solid [16, 17]. Understanding the supramolecular self-assembly of molecules via intermolecular interaction tends to explain the relationship between the molecular structure and the properties of the crystalline material [18, 19]. Interestingly in the case of acridinium/acridine derivative crystals, the formation of π…π and C‒H…π interaction between the acridine planes significantly influences the luminescence property of the material [20, 21]. Owing to these interests, we intend to study the crystal structure, supramolecular features, photophysical properties, and thermal properties of three acridinium derivatives and one acridine compound which is presented in this paper.
In this present work, we adopted a mechanochemical synthesis (liquid-assisted grinding) procedure in the preparation of new acridinium/acridine derivatives. In recent years, the concept of mechanochemical synthesis has attracted researchers to prepare simple organic compounds/derivatives due to their various significance (higher yield, solvent-free, environment friendly, etc.) over conventional chemical synthesis procedures [22,23,24,25]. The details of synthesis, structural studies, and other characterizations of 2-(acridinium-9-yl oxy) phenol chloride (I), 4-(acridinium-9-yl oxy) chlorobenzene chloride (mono)chloroform solvate (II), 4-(acridinium-9-yl) naphthalene-1,3-diol chloride (mono)ethanol solvate (III), and 2-(acridin-9-yl) naphthalene 1,8-diol (IV) derivatives are presented in the preceding section.
Experimental
Synthesis and crystallization
All acridine-based derivatives (I–IV) discussed in this paper were prepared by mechanochemical synthesis. The mixture of 9-chloroacridine or acridine and selected compounds was subjected to solvent-assisted grinding using an agate mortar and pestle. The ground powder was then dissolved in a suitable solvent for crystallization as illustrated in Fig. 1. The FT-IR spectra of the derivatives (I) to (IV) are given in the Supporting Information Figs. S1–S4.

Illustrates the steps involved in the mechanochemical synthesis of compounds
I. 2-(Acridinium-9-yl-oxy) phenol chloride [C19H14NO2 +Cl−]
Initially, equimolar quantities of 9-chloroacridine (0.213 g/1.0 mM) and 1,2-dihydroxybenzene (0.110 g/1.0 mM) were taken and ground using an agate mortar. CHCl3 solvent was added dropwise and ground to a fine paste, and the grinding was continued in three steps for 15 min until a change in color of the mixture was noticed. The resultant powder was then dissolved in a mixed solvent of dichloromethane and chloroform (50:50) followed by continuous stirring for 20 min until a clear solution was attained. Thus, a prepared clear solution of compound (I) was kept for slow evaporation without any atmospheric disturbance. Good diffraction quality yellow-colored crystals of (I) were obtained after 10 days. FT-IR studies: O‒H = 3332 cm−1, 3198 cm−1, C‒H = 3050 cm−1, C=N = 1885 cm−1, N–H = 1637 cm−1, C=C = 1580 cm−1, C–H = 1408 cm−1, O‒H = 1367 cm−1, C‒N = 1270 cm−1, C‒O = 1112 cm−1, C=C = 945 cm−1, C‒H = 850 cm−1, 751 cm−1.

II. 4-(Acridinium-9-yl-oxy) chlorobenzene chloride (Mono)chloroform solvate [C19H13NOCl+Cl−. CHCl3]
Equimolar quantities of 9-chloroacridine (0.213 g/1.0 mM) and 4-chlorophenol (0.110 g/1.0 mM) were mixed and ground using an agate mortar. CHCl3 solvent was added dropwise and ground to a fine paste, and the grinding was continued in three steps for 10 min until a color change in the mixture occurred. The resultant powder was then dissolved in a mixed solvent of dichloromethane and chloroform (50:50) and stirred continuously for 15 min to attain a clear solution. Thus, a prepared clear solution of compound (II) was kept for slow evaporation without any atmospheric disturbance. Good diffraction-quality yellowish orange crystals of (II) were obtained after 1 week. FT-IR studies: O‒H = 3175 cm−1, C‒H = 3081 cm−1, C=N = 1882 cm−1, N–H = 1628 cm−1, C=C = 1582 cm−1, C–H = 1364 cm−1, O‒H = 1364 cm−1, C‒N = 1314 cm−1, C‒O = 1086 cm−1, C=C = 959 cm−1, C‒Cl = 827 cm−1, C–H = 867 cm−1, 751 cm−1.

III. 4-(Acridinium-9-yl) naphthalene-1,3-diol chloride (Mono)ethanol solvate [C23H22NO2 +Cl−. C2H5OH]
Equimolar quantities of 9-chloroacridine (0.213 g/1.0 mM) and 1,3-dihydroxynaphthalene (0.160 g/1.0 mM) were taken and ground using an agate mortar. Dichloromethane solvent was added dropwise and ground to a fine paste, and the grinding was continued in three steps for 10 min until a change in color of the mixture was noticed. The resultant powder was then dissolved in a mixed solvent of dichloromethane and ethanol (50:50) and stirred continuously for 20 min to attain a clear solution. Thus, a prepared clear solution of compound (III) was kept for slow evaporation without any atmospheric disturbance. Good diffraction-quality purple-colored crystals of (III) were obtained after 10 days. FT-IR studies: O‒H = 3140 cm−1, C‒H = 3049 cm−1, C=N = 1844 cm−1, N–H = 1627 cm−1, C=C = 1574 cm−1, C–H = 1395 cm−1, O‒H = 1362 cm−1, C‒N = 1310 cm−1, C‒O = 1236 cm−1, C=C = 987 cm−1, O‒H = 929 cm−1, C‒H = 850 cm−1, 751 cm−1.

IV. 2-(Acridin-9-yl) naphthalene 1,8-diol [C23H15NO2]
Initially, equimolar quantities of acridine (0.179 g/1.0 mM) and 1,8-dihydroxynaphthalene (0.160 g/1.0 mM) were ground using an agate mortar as followed in previous compounds. Dichloromethane solvent was added dropwise and ground to a fine paste, and the grinding was continued in three steps for 15 min until a color change in the mixture occurred. The resultant powder was then dissolved in a mixed solvent of dichloromethane and ethanol (50:50) followed by continuous stirring for about 25 min till a clear solution (dark blue) was attained. Thus, a prepared clear solution of compound (IV) was kept for slow evaporation without any atmospheric disturbance. Good diffraction-quality dark blue-colored crystals of (IV) were obtained after 2 weeks. FT-IR studies: O‒H = 3170 cm−1, C‒H = 3049 cm−1, C=N = 1811 cm−1, C=C = 1599 cm−1, O‒H = 1362 cm−1, C‒N = 1300 cm−1, C‒O = 1231 cm−1, O‒H = 908 cm−1, C‒H = 843 cm−1, 678 cm−1.

Data collection and refinement
Good diffraction quality crystals of (I) to (IV) were obtained from the crystallization process, of which suitable sizes (see Table 1) of crystals were chosen for the X-ray diffraction experiment. Intensity data of the crystals were collected using Bruker D8 Venture Photon II Diffractometer equipped with a micro-focus dual source sealed tube. The intensity data for compounds (I) and (III) were collected using Mo-source (λ = 0.7103 Å) whereas the intensity data for compounds (II) and (IV) were collected using Cu source (λ = 1.54178 Å), respectively. Correct unit-cell parameters were determined by collecting data from 60 frames in three different matrices followed by the data collection, the exposure time of 10 s per frame, and a scan width of 0.5˚. Data reduction, scaling, and multi-scan absorption correction were carried out using SAINT-plus and SADABS in APEX3 software [26]. The crystal structures were solved by direct methods procedure using SHELXS [27] program and refined by full-matrix least square procedure on F2 using the SHELXL-2018 [27] program. The intensity data was checked for missing symmetry elements and Twinng with PLATON program [28]. In the crystal structure of (III), the solvent ethanol molecule is positionally disordered over two positions with equal site occupancies of 0.5:0.5. Hydrogen atoms were placed in geometrically idealized positions and constrained to ride on their parent atoms with C‒H distances in the range 0.93‒0.97(Å). Isotropic thermal parameters (Ueq) were fixed such that Uiso(H) = 1.5Uequ(C) for methyl C‒H bond and Uiso(H) = 1.2Uequ(C) for methylene and aromatic C‒H bond, respectively. The hydroxyl hydrogen atoms were identified from the difference electron density map and allowed to ride on the parent oxygen atom. The molecular graphic images of the molecules were drawn using OLEX-2 and Mercury 3.9 [29, 30] software. The crystal structure refinement details of (I) to (IV) are summarized in Table 1. The finalized structure refinement files are deposited in the Cambridge Crystallographic Data Centre (https://ccdc.cam.ac.uk), and the data files of compounds (I)‒(IV) can be accessed using the Id CCDC2357373, 2357374, 2357375, and 2357376, respectively.
Result and discussion
Crystal structure and supramolecular features
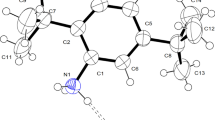
The molecular structure of the acridinium and acridine compounds (I) to (IV) showing the displacement ellipsoids is depicted in Fig. 2. These compounds exhibit different types of intermolecular interactions responsible for the formation of supramolecular frameworks. The details of the intermolecular interaction geometry of hydrogen bonds and other weak interactions are given in Tables 2, 3, 4, and 5.

Molecular structure of compounds (I)–(IV) showing the displacement ellipsoids drawn at a 50% probability level
2-(Acridinium-9-yl-oxy) phenol chloride (I)
Compound (I) crystallizes in monoclinic spacegroup P21/n with one acridinium cation [2-(acridinium-9-yl oxy) phenol] and one chloride anion in the asymmetric unit. In the molecular structure, the phenyl plane of the 1,2-dihydroxybenzene moiety deviates from the planar acridinium moiety with a dihedral angle of 75.65(9)˚ The bridging oxygen atom between the phenol group and the acridinium moiety makes an angle of 120.19(12)˚, respectively. In the crystal structure, the inversion-related acridinium cation and its Cl− anions are interlinked through N‒H…Cl and O‒H…Cl hydrogen bonds such as N1‒H1A…Cl1 and O2‒H2A…Cl1ii, respectively, constitute the basic super-molecule. The adjacent super-molecules are further linked through a C19‒H19…Cl1i interaction, thus generating a one-dimensional supramolecular tape extending infinitely along the crystallographic [1 0 0] direction as shown in Fig. 3a. Interestingly in the one-dimensional tape, the Cl− anions act as a trifurcated acceptor for different types of interactions, resembling a knot which tides the acridinium cations in the supramolecular framework. The [1 0 0] supramolecular tapes stacked along the b-axis are further interlinked through a C‒H…Cl interaction such as C7‒H7…Cl1 resulting in the formation of a two-dimensional supramolecular sheet extending parallel to the (0 0 1) plane as depicted in Fig. 3b. A weak C‒H…O and C‒H…π interactions such as C2‒H2…O2 and C3‒H3…π4iii connect the adjacent two-dimensional supramolecular (0 0 1) sheet resulting in the formation of a three-dimensional network, which constitutes the molecular packing in the crystalline solid.

Part of the crystal structure of (I) showing a the formation of one-dimensional supramolecular tape extending parallel to crystallographic [1 0 0] direction and b the formation of two-dimensional supramolecular sheet extending parallel to (0 0 1) plane. Hydrogen atoms not involved in intermolecular interactions are omitted for clarity
4-(Acridinium-9-yl oxy) chlorobenzene chloride (Mono)chloroform solvate (II)
The compound (II) crystallizes in monoclinic spacegroup P21/c with one acridinium cation [(4-(acridinium-9-yl oxy) chlorobenzene], one chloride anion, and a chloroform solvate in the asymmetric unit. In the molecular structure, the phenyl plane of the chlorophenol moiety deviates significantly from the acridinium plane with a dihedral angle of 83.94(5)˚ while the bridging oxygen atom connecting the chlorophenyl moiety and the acridinium moiety makes a C‒O‒C angle of 118.7(3)˚, respectively. In the crystal structure, the inversion-related acridinium cation and the Cl− anions are interlinked through N‒H…Cl and C‒H…Cl hydrogen bonds such as N1‒H1A…Cl1 and C15‒H15…Cl1i, respectively, constitute the basic super-molecule. The adjacent super-molecules are interlinked through C6‒H6…Cl1 interaction forming a one-dimensional supramolecular tape extending parallel to the crystallographic [1 0 0] direction as depicted in Fig. 4a. Parallel [1 0 0] supramolecular tapes are further bridged by the chloroform solvent molecule via. C‒H…Cl interactions generate a three-dimensional network in the crystal structure. The bridging chloroform solvent forms C20‒H20…Cl1i interaction with the chloride anion and C20‒H20…Cl2ii interaction with the chlorine atom of the acridinium cation, respectively. Interestingly, in the three-dimensional network, the chloroform solvent molecules are connected by Cl…Cl halogen bonds such as Cl3…Cl5iii and Cl4…Cl5iii forming a zig-zag solvate chain extending along the [0 1 0] direction (Fig. 4b). It resembles that the chloroform solvent chain is trapped between the acridinium, Cl− molecular network channels, mimics a host-guest type of architecture in the three-dimensional crystal structure as shown in Fig. 4b.

a The formation of one-dimensional supramolecular tape extending parallel to crystallographic [1 0 0] direction and b the formation of a three-dimensional molecular network bridged via CHCl3 solvent chain in the crystal structure of (II). Hydrogen atoms not involved in intermolecular interactions are omitted for clarity
4-(Acridinium-9-yl) naphthalene-1,3-diol chloride (Mono)ethanol solvate (III)
The compound (III) crystallizes in triclinic spacegroup P-1 with one acridinium cation [4-(acridinium-9-yl) naphthalene-1,3-diol], one chloride anion, and an ethanol solvent molecule in the asymmetric unit. In the molecular structure, the naphthalene plane makes a deviation from the acridinium plane with a dihedral angle of 69.53(8), the C‒C bond length which connects the naphthalene moiety, and the acridinium moiety is found to be a value of 1.484(3)Å. In the crystal structure, the adjacent acridinium cations are knotted by the chlorine atom via N‒H…Cl and O‒H…Cl hydrogen bonds such as N1‒H1B…Cl1 and O1‒H1A…Cl1 forming a one-dimensional supramolecular chain extending parallel to the crystallographic [1 −1 0]. Interestingly, the inversion-related adjacent one-dimensional supramolecular chains are bridged by the lattice ethanol solvent molecule via O2‒H2A…O3 and O3‒H3A…Cl1ii hydrogen bonds resulting in the formation of one-dimensional supramolecular tape propagating along crystallographic [1 −1 0] direction [Fig. 5a]. The adjacent supramolecular tape of the type [1 −1 0] is further interlinked through a C‒H…O interaction such as C1‒H1…O1 which generates a two-dimensional supramolecular sheet spreading parallel to the (1 1 1) plane as depicted in Fig. 5b. These two-dimensional sheets are additionally stabilized by the presence of π…π interaction exhibited between the acridinium planes.

Part of the crystal structure of (III) showing a the formation of one-dimensional supramolecular tape bridged by CH3OH solvent extending along the crystallographic [1 − 1 0] direction and b the formation of two-dimensional supramolecular sheet extending parallel to (1 1 1) plane. Hydrogen atoms not involved in intermolecular interactions are omitted for clarity
2-(Acridin-9-yl) naphthalene-1,8-diol (IV)
The acridine compound (IV) crystallizes in monoclinic spacegroup P21/n with four molecules in the unit cell. In the molecular structure, the naphthalene plane and the acridine plane deviate significantly from each other with an observed dihedral angle of 76.28˚(9). The C‒C bond length which connects the naphthalene moiety and the acridine moiety is found to be a value of 1.497(3)Å, which is slightly longer than that observed in the acridinium cation of compound (III). It is observed that in the dihydroxynaphthalene moiety, one of the hydroxyl groups (O1‒H1A) forms an intramolecular O‒H…O hydrogen bond with the neighboring hydroxyl group (O2‒H2A) oxygen atom whereas the hydroxyl group O2‒H2A forms an intermolecular O‒H…N hydrogen bond with the neighboring molecule. In the crystal structure, the adjacent acridine molecules are linked through strong O2‒H2A…N1i hydrogen bonds which generate a one-dimensional supramolecular chain extending parallel to the crystallographic [1 0 −2] direction as shown in Fig. 6a. The adjacent [1 0 −2] chains are further interlinked through C‒H…O and C‒H…π interactions such as C6‒H6…O1ii and C2‒H2…π1iii, respectively, which results in the formation of two-dimensional supramolecular grid type architecture spreading parallel to the (0 1 0) plane and is depicted in Fig. 6b. In the two-dimensional supramolecular architecture, the adjacent one-dimensional [1 0 −2] chains are separated by a distance of 6.617 Å with respect to the naphthalene planes. Similarly, the adjacent acridine planes in the two-dimensional grid are inclined to each other with a calculated dihedral angle of 65.43˚. The three-dimensional molecular packing in the crystal is further stabilized by the presence of weak C‒H…N and π…π interactions.

Part of the crystal structure of (IV) showing a the formation of a one-dimensional supramolecular chain linked via O–H…N hydrogen bond extending along the crystallographic [1 − 1 0] direction and b the formation of a two-dimensional supramolecular sheet extending parallel to (1 1 1) plane. Hydrogen atoms not involved in intermolecular interactions are omitted for clarity
π–π Interaction between acridinium/acridine moieties
The significant feature as far as the acridinium/acridine molecules are concerned is their antiparallel π…π stacking interactions with the neighboring molecules, which helps to understand various properties of the compound in the solid-state [20]. We intend to understand the acridine-acridine stacking via π-contacts in the three-dimensional crystal structure. The geometries of π-contacts for compounds (I)‒(IV) are given in Tables 2, 3, 4, and 5. In the crystal structure of (I), the adjacent acridine moieties are stacked in a parallel-displaced manner through π1…π1ii and π2…π3ii contact as shown in Fig. 7a with a distance of 3.8300(6)Å between the acridine planes. The adjacent acridinium moieties are as follows slipped to a distance of 1.567 Å with a deviation angle of 24.5˚, respectively. In the crystal structure of (II), the adjacent acridine moieties deviate away from the expected π…π contact geometry with a centroid-to-centroid distance of 4.561(2)Å, and this deviation is mainly attributed to the formation of an O…π contact between the oxygen atom O2 and the phenyl plane Cg(C9/C10/C11/N1/C12/C13) with an observed distance of O2…π1iv = 3.151(3) Å (Fig. 7b). Moreover, a Cl…π contact is observed between the perchlorate chlorine atom Cl3 and the centroid of the benzene plane Cg(C5/C6/C7/C8/C13/C12), [Cl3…π2v = 3.308(3) Å]. These O…π and Cl…π contact geometry are in agreement with the previously reported structures [31]. The inversion-related acridine moieties in the crystal structure of (III) deviate from each other such that a π…π contact is observed between the phenyl planes C5/C6/C7/C8/C13/C12 with a separation distance of π1…π1ii = 3.647(3) Å as shown in Fig. 6c. This slip in the acridine dimer is attributed to the presence of solvent methanol molecule in the lattice, which potentially forms strong interaction between the acridinium cation and Cl− anion, respectively. In the crystal structure of (IV), as observed in (II), the adjacent acridine moieties deviate away from the expected π…π contact distance with a centroid-to-centroid distance of 4.001(9)Å, and this deviation can be justified by the formation of a weak C‒H…N interaction (C16‒H16…N1v) formed between the inversion related molecules and is shown in Fig. 7d. In general, from the crystal structures of (I) to (IV), the antiparallel π…π stacking interactions between acridinium or acridine moieties deviates from the expected geometry because of the steric hindrance induced by the substituents and the intermolecular interaction formed with the solvent molecule in the lattice.

a‒d The probable π…π contacts observed between the acridinium/acridine moieties in the crystal structures of (I) to (IV). Hydrogen atoms not involved in interactions are omitted for clarity
Hirshfeld surface analysis
To gain a better understanding of the non-covalent interactions and their percentage contribution to the crystal, the Hirshfeld surface analysis was performed for all crystal structures using Crystal Explorer 21.5 software [32]. The normalized contact distance (dnorm) based on the internal (di) and external (de) distances to the nearest nucleus and van der-Waals radii of atoms is given by dnorm = (di-rivdw/rivdw)⁄(de-revdw/revdw), where rivdw and revdw are the van der Waals radii of the appropriate atoms internal and external to the surface [33]. This enables the identification of regions of particular importance to intermolecular interactions. Moreover, the combination of di and de represented in the form of a Fingerprint plot provides a summary of intermolecular interaction in the crystal.
2-(Acridinium-9-yl oxy) phenol chloride (I)
The Hirshfeld Surface (HS) and the corresponding two-dimensional fingerprint (FP) plot of (I) have been mapped over the dnorm of −0.6075 to 1.5282 and are depicted in Fig. 8a. The HS shows that the intense red spots are due to the presence of strong N‒H…Cl and O‒H…Cl hydrogen bonds. The faint red spots around the C‒H atom are due to the presence of C‒H…Cl interaction whereas the intense white region around the C3 atom is attributed to the formation of C‒H…π interaction observed in the crystal structure. Similarly, the HS over the acridine moiety shows uniformly scattered white regions corresponding to the presence of π…π interaction between the acridine planes. The two-dimensional fingerprint plot delineates into H…H, H…C/C…H, H…O/O…H, H…Cl/Cl…H, C…C, N…C/C…N contacts, and their relative contributions are illustrated in Fig. S5 (supporting information). To the overall packing the most important contact originates from H…H interaction with a contribution of 44.1% as widely scattered points in the fingerprint plot with the tip at de + di ≈ 2.2 Å. The H…C/C…H contact representing the C‒H…π interaction contributes 28.1% showing a pair of broad spikes at de + di ≈ 2.8 Å. A pair of characteristic wings in the FP plot delineate into H…O/O…H contact represents the presence of C‒H…O interaction, viewed as a pair of spikes terminating at de + di ≈ 2.55 Å with a contribution of 9.5%. A sharp spike with the tip at de + di ≈ 2.0 Å is attributed to the presence of H…Cl/Cl…H contact contributes 8.2% to the molecular packing. The sharp green spots over the sharp blue spike represent the presence of N‒H…Cl hydrogen bonds in the crystal structure. The C…C contacts representing the π…π interaction between the acridinium moieties contribute 6.1% showing bullet-shaped distributed points with concentrated green spots terminating at de + di ≈ 3.4 Å. Other weak contacts C…N, C…Cl and H…N together contribute 3.2% to the overall molecular packing.

a–d Hirshfeld surface mapping and the corresponding two-dimensional fingerprint plot of compounds (I) to (IV)
4-(Acridinium-9-yl oxy) chlorobenzene chloride (Mono)chloroform solvate (II)
For the compound (II), the HS analysis and two-dimensional fingerprint plot have been mapped over dnorm of −0.6042 to 1.1079 and is shown in Fig. 8b. In the HS map of (II), the intense red spot is attributed to the presence of N‒H…Cl hydrogen bond whereas the faint red spots over the surface are due to the presence of C‒H…Cl interaction was observed in the crystal structure. The HS over the acridinium moiety shows some intense blue region is attributed to the absence of C…C contacts or any significant π…π interaction between the acridine fragment and is clear from the crystal structure description. The two-dimensional fingerprint plot of (II) is delineated into H…Cl/Cl…H, H…H, H…C/C…H, Cl…C/C…Cl, C…O/O…C, H…N/N…H, Cl…Cl, and C…C is shown in Fig. S6. The most important contact originates from H…Cl/Cl…H contacts contribute 31.4% to the overall packing of the crystal representing the presence of N‒H…Cl and C‒H…Cl interactions. In the two-dimensional fingerprint plot, the intense green spots over the blue spike with the tip at de + di ≈ 2.0 Å are due to the presence of N‒H…Cl hydrogen bond whereas the broad spike with scattered blue points at de + di ≈ 2.61 Å is due to the presence of C‒H…Cl interactions in the crystal structure. The widely scattered points in the fingerprint plot due to the H…H contact contribute 29.1% to the molecular packing showing a spike at de + di ≈ 2.36 Å. In the absence of significant C‒H…π interactions in the crystal structure, the H…C/C…H contact shows a pair of broad spikes with the tip at de + di ≈ 2.8 Å contributing about 22.9% in the molecular packing. The Cl…C/C…Cl contact representing the Cl…π interaction in the crystal structure contributes 7.6% showing a bullet-shaped distribution of points in the fingerprint plot terminates at de + di ≈ 3.28 Å. The O…C/C…O contact representing the O…π interaction in the crystal structure contributes 3.3% showing some scattered points in the center of the fingerprint plot. Other contacts such as H…N/N…H, Cl…Cl, and C…C together contribute 5.0% to the overall molecular packing of the crystal.
4-(Acridinium-9-yl) naphthalene-1,3-diol chloride (Mono)ethanol solvate (III)
The HS and two-dimensional fingerprint plot for compound (III) has been mapped over the dnorm of −0.7505 to 1.2937 and is depicted in Fig. 8c. The HS map of (III) shows that the intense red spots are attributed to the presence of strong N‒H…Cl, O‒H…Cl and O‒H…O hydrogen bonds. The faint red spots observed on the surface are due to the presence of weak C–H…O interaction in the crystal structure. The uniform white surface over the phenyl plane C5/C6/C7/C8/C13/C12 (of acridine fragment) is due to the presence of π1…π1 interaction observed in the crystal structure. The two-dimensional fingerprint plot of (III) delineated into H…H, H…C/C…H, H…O/O…H, C…C, H…Cl/Cl…H, H…N/N…H, C…O/O…C, and N…C/C…N is depicted in Fig. S7. The major contribution to the overall packing of the crystal is from H…H contact showing 47.9% observed as widely scattered points over the fingerprint plot with the tip at de + di ≈ 2.07 Å. The H…C/C…H contact contributes 26.5% to the overall packing representing C‒H…π interaction in the crystal structure showing a pair of characteristic spikes terminating at de + di ≈ 2.60 Å and 2.73 Å, respectively. It is observed that the C…C contact representing the π…π interaction contributes 6.8% to the molecular packing corresponds to concentrated green spots over blue points in the center of the two-dimensional plot at de + di ≈ 3.4 Å. The scattered points that terminate with a sharp spike at de + di ≈ 3.4 Å are due to the presence of H…Cl/Cl…H contact contributing 5.8% to the overall packing. The green dotted spots over the blue spike are attributed to the formation of N‒H…Cl hydrogen bonds in the crystal structure. Other weak contacts H…N/N…H, C…O/O…C, and N…C/C…N together contribute 2.8% to the overall molecular packing in the crystal.
2-(Acridin-9-yl) naphthalene 1,8-diol (IV)
The HS and two-dimensional fingerprint plot for compound (IV) has been mapped over the dnorm of −0.7219 to 1.6117 and is depicted in Fig. 8d. As observed in other structures, the intense red spot on the HS is attributed to the formation of a strong O‒H…N hydrogen bond whereas the faint red spot is due to the presence of C‒H…O and C‒H…π interactions in the crystal structure. The two-dimensional fingerprint plot delineated into H…H, H…C/C…H, H…O/O…H, C…C, H…N/N…H, and N…C/C…N contacts, and their relative contributions are illustrated in Fig. S6. The most important contact in the overall packing originates from H…H interaction with a contribution of 47.6% as widely scattered points in the fingerprint plot with the tip at de + di ≈ 2.02 Å. The H…C/C…H contact representing the C‒H…π interaction contributes 27.9% and is depicted as a pair of broad spikes terminating at de + di ≈ 2.52 Å. A pair of characteristic wings in the FP plot delineate into H…O/O…H contact represents the presence of C‒H…O interaction viewed as a pair of spikes with the tip at de + di ≈ 2.51 Å with a contribution of 9.2%. to the molecular packing. Though there is no significant strong π…π interaction observed between the acridine moieties in the crystal structure, the fingerprint plot shows that the C…C contacts contribute 8.9% represented as bullet-shaped points with concentrated green spots terminating at de + di ≈.
3.7 Å. A pair of sharp spikes with the tip at de + di ≈ 2.0 Å is attributed to the presence of H…N/N…H contact representing the O‒H…N hydrogen bond contributes 6.1% to the overall molecular packing in the crystal structure.
Molecular electrostatic potential
The molecular electrostatic potential (ESP) for the compounds (I) to (IV) were calculated by adopting HF/STO-3G wavefunction using Crystal Explorer 21.5 software. The corresponding electrostatic potential over the molecular surface has been calculated in the energy range of −0.05 to 0.05 a.u. and is illustrated in Fig. 9, and the blue region around the atom corresponds to the positive potential whereas the red region corresponds to the negative potential. From the electrostatic potential map, it is observed that the formation of strong negative potential around the Cl− anion enlarged over a certain region for the acridinium molecules (I) to (III) indicates that the Cl− atom acts as a potential acceptor moiety in crystal packing. This can be justified from the crystal structure by the formation of various Cl− anion-assisted hydrogen bonds (N‒H…Cl and O‒H…Cl), weak interactions (C‒H…Cl), and halogen bonds (Cl…Cl) (see Tables 2, 3, 4, and 5). In the molecular structure of (I) and (II), the negative electrostatic potential of bridging oxygen atom O1 looks diminished compared to the hydroxyl oxygen atom O2 (I). This is due to the situation that the bridging oxygen atom O1 does not participate in any strong interaction compared to the hydroxyl oxygen atom O2.

Electrostatic potential isosurface calculated at 0.05 a.u. of compounds (I) to (IV). The electrostatic potential calculated for compounds (I) to (III) without Cl− anion is shown in Fig. S9
In the ESP map of (IV), it is observed that there exists a significant negative potential over the acridine nitrogen atom (N1), which acts as an acceptor atom for the formation of a strong O‒H…N hydrogen bond in the crystal structure. It is evident that a negative potential surface exists around the oxygen atom O1 whereas a positive potential forms around the oxygen atom O2. This can be attributed to the fact that the oxygen atom O1 acts as an acceptor atom in the formation of C6‒H6…O1 interaction whereas the oxygen atom O2 acts as a donor atom in the formation of strong O2‒H2A…N1 hydrogen bond, respectively.
Interaction energy studies
To understand the stability of the crystal, we intend to calculate the interaction energy between the molecular pairs for the cocrystals (I) to (IV) using the program, Crystal Explorer 21.5. The interaction energy between the molecular pairs has been calculated using dispersion-corrected HF-31G (d, p) quantum level theory. The calculated total energy (Etot) is the sum of other energy components such as electrostatic energy (Eele), polarization energy (Epol), dispersive energy (Edis), and exchange-repulsion energy (Erep), respectively [34, 35]. The details of scale factors used for calculation and the interaction energy values for cocrystals (I) to (IV) are given in the program output Tables S1 to S4 (supporting information).
The intermolecular interaction energy calculation between the molecular pairs in compound (I) shows that the total energy of charge-assisted hydrogen bonds N1‒H1A…Cl1/O2‒H2A…Cl1 is observed to be −29.8 kJmol−1. The total energy between the molecular pair for C3–H3…π4 interaction is calculated to be −14.4 kJmol−1, which is slightly stronger than other weak interactions such as C9‒H9…Cl1 [−7.6 kJmol−1], C7‒H7…Cl1 [−11.8 kJmol−1] and C2‒H2…O2 [−8.9 kJmol−1], respectively. Interestingly, the C‒C contact exhibited between the acridinium moieties representing the formation of π1…π1ii and π2…π3ii interactions show a calculated total energy value of −36.8 kJmol−1, which is stronger compared to other intermolecular interactions (Fig. 10a).

a–d Calculated total interaction energy values exhibited between the molecular pairs in the crystal structure of compounds (I) to (IV)
In the molecular structure of (II), from the interaction energy calculation, it is observed that the total energy of charge-assisted hydrogen bonds N1‒H1A…Cl1/C15‒H15…Cl1 is observed to be −27.6 kJmol−1. The energy value of C‒H…Cl interaction formed with respect to the Cl− anion is observed to be −16.9 kJmol−1 (C6–H6…Cl1) and −11.6 kJmol−1 (C20‒H20…Cl1) which is slightly stronger than the C20‒H20…Cl2 (−9.1 kJmol−1) and C16‒H16…Cl4 (−9.1 kJmol−1) interactions formed between the CHCl3 solvate and the substituted Cl atom. Interestingly, the strength of O2…π1/π…π contact exhibited between the inversion-related acridinium molecular pairs is significantly dominant over other interactions with a total energy value of −36.6 kJmol−1 (Figs. 7b and 10b).
The interaction energy between the molecular pairs in compound (III) shows that the total energy associated with the Cl− acceptor atom signifying the hydrogen bonds N1‒H1B…Cl1, O1‒H1A…Cl1 is observed to be −27.9 kJmol−1. It is of interest to note that the total energy calculated between the molecular pairs forming O2‒H2A…O3 and O3‒H3A…Cl1 hydrogen bonds show a value of −45.2kJmol−1 which is comparatively stronger than all other interactions (Fig. 10c). This strong energy value can be due to the nature of hydrogen bonds formed between strong electronegative O and Cl− atoms (Table 4). The total energy calculated for π…π interaction between the acridinium molecular planes shows a value of −25.9 kJmol−1, which is weaker compared to the energy value of π…π interaction observed in compounds (I) and (II). It is clear that in (III), the π…π stacking of acridinium moieties shows that the C…C contact is exhibited between only one terminal phenyl plane (see Fig. 7c).
From the interaction energy calculation of compound (IV), it is observed that the O2‒H2A…N1 hydrogen bond is the strongest with a total energy value of −72.3 kJmol−1 (Fig. 10d). The calculated energy value of C‒H…π interactions exhibited between the molecular pairs is found to be −25.3 kJmol−1 (C2‒H2…π1) and −31.3 kJmol−1 (C18‒H18…π2), whereas the total energy value of the C6–H6…O1 is calculated to be −18.9 kJmol−1 which is comparatively weaker than the C‒H…π interactions. As observed in other structures, the energy value between the acridine molecular pairs representing the π…π contact along with C16‒H16…N1 interaction is calculated to be −42.7 kJmol−1, which is significantly stronger than other weak interactions.
Photophysical properties
UV–visible and P L studies
To gain an understanding of the photophysical properties of the molecules, we conducted UV-visible absorption and photoluminescence spectroscopy under highly dilute conditions (~12 µM) to mitigate the impact of self-association. The absorption and emission spectra of acridinium derivatives (I), (II), and (III) were recorded in chloroform solvent, whereas for acridine compound (IV), the spectra were recorded on ethanol solvent and are shown in Figs. 11 and 12. The absorption spectra illustrate that all four molecules exhibit a characteristic broad absorption band in the range of 320 to 420 nm, reminiscent of acridine, with discernible peaks indicating electronic transitions. Remarkably, the UV-visible spectra displayed prominent peaks corresponding to π–π* transitions within acridine’s tricyclic aromatic structure, providing crucial insights into the energy levels associated with electronic excitations and contributing to a comprehension of acridine moiety’s absorption behavior [36]. These broad, structureless features for (I), (II), and (III) well resemble previously reported acridinium salt absorption features [37], whereas (IV), which does have a neutral acridine chromophore, is quite different from others [38]. It is also worth mentioning that the additional broad feature between 360 and 400 nm seen in the case of (I) and (II) is due to the presence of the ether linkage, which allows the molecules to have more degrees of freedom and more perturbation. Moreover, ethers may not exhibit strong absorption in the UV range, but they can affect the electronic transitions of adjacent chromophores (acridine here), influencing the overall electronic structure of the molecules, and potentially leading to a hypochromic shift in absorption maxima. Notably, for (III) and (IV), an additional structured absorption band can be seen with characteristic peaks at 380, 360, and 340 nm, well-documented as the naphthalene absorption band in the literature [39].

Absorption spectra of compounds (I) to (IV)

Emission spectra of compounds (I) to (IV)
Subsequently, we analyzed the photoluminescence (PL) spectra to elucidate the fluorescence properties of acridine upon excitation at the corresponding absorption maxima (I at 380 nm, II at 370 nm, III at 395 nm, and IV at 388 nm). The PL spectra exhibited characteristic emission peaks falling within the 400 to 500 nm range, with structural features mirroring the acridine absorption bands. Each emission peak corresponds to specific electronic transitions identified in the absorption spectra. The visible range fluorescence emission not only demonstrated the efficient relaxation of excited states but also provided valuable insights into the photophysical behavior of acridinium/acridine chromophores.
Unlike acridinium derivatives (I, II, and III), the acridine compound (IV) exhibited considerable fluorescence quenching, which is attributed to donor-acceptor charge transfer interactions where the lone pairs of nitrogen in the acridine moiety increase the donating capacity of acridine donor to the naphthalene acceptor [40]. This charge transfer transition is utterly absent in (I) and (II) due to the unavailability of donating lone pairs, resulting in higher PL emissions. However, a moderate lowering in PL emission is observed in the case of (III), where although N lone pair is not available, the protonated acridine moiety can still act as a donor moiety, similar to anthracene donor and operate a D-A transition to the high electron deficiency of naphthalene acceptor unit [41]. To support our findings from PL emissions, we have calculated the PL quantum yield for all these emissions, taking Rhodamine-B as a reference. The quantum yield for (I) is 0.47, and for (II) is 0.44, which faces a huge drop to 0.07 for (IV) as the result of PL quenching due to charge transfer interactions. Meanwhile, the acridinium compound (III) exhibits a lower PL quantum yield of 0.13, supporting the partial quenching due to charge transfer experienced within the molecule. The correlation between absorption and PL spectra offers a comprehensive perspective on the electronic structure and photoluminescent characteristics of acridine moiety, opening avenues for its application in diverse fields, ranging from materials science to fluorescence probing in biological systems.
Thermogravimetric analysis
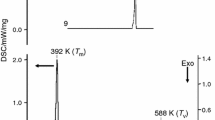
Thermogravimetric analysis (TGA) was carried out using the TA instrument Q500 model. The samples were equilibrated and kept in isotherm at 30 °C for 2 min. The thermogravimetric analysis of compounds (I)‒(IV) was carried out from room temperature to 500 °C for compounds (I)‒(III) and up to 1000 °C for compound (IV). Compounds (I)‒(III) degrade in three steps whereas compound (IV) undergoes a single-step degradation and is depicted in Fig. 13.

TGA plot of compound (I) to (IV)
In compound (I), the first step of thermal degradation that occurs in the temperature range of 100‒200 °C is attributed to the loss of chlorine ions from the lattice. The second step of degradation observed in the temperature range of 200‒300 °C is due to the loss (or break up) of the 1,2-dihydroxyphenol fragment. The third step of degradation is observed in the temperature range of 360‒450 °C is attributed to the loss of acridine fragment or disintegration of a complete molecule. The degradation plot of temperature vs weight loss for compound (I) is shown in Fig. 5(I).
From the TGA plot of compound (II), the thermal degradation observed in the temperature range of 50‒100 °C is attributed to the loss of solvent molecule CHCl3 in the lattice. The second step of thermal degradation observed between the temperature range 100‒190 °C is due to the loss of the chlorine atom in the crystal. The third stage of degradation is attributed to the loss (or breakup) of the 4-chlorophenol fragment of the molecule, observed in the temperature range of 190‒300 °C. The final stage of degradation observed in the temperature range 300‒350 °C is due to the breakdown of the acridine moiety of the molecule, 20% of the molecular fragment or carbon atom is left as residue beyond 350 °C. The degradation plot of temperature vs weight loss for compound (I) is shown in Fig. 5(II).
Compound (III) undergoes the first step of thermal degradation in the temperature range of 110–190 °C is attributed to the loss of methanol solvent and chlorine ion in the lattice. Interestingly, no expected weight loss is observed in the temperature range 190‒265 °C, followed by the second stage thermal degradation of the compound was observed in the temperature range 265–390 °C is attributed to the loss (or breakdown) of the 1,3-dihydroxynaphthalene fragment in the molecule. The final stage of degradation takes place in the temperature range of 390‒450 °C and is expected to the breakdown of the acridine moiety of the molecule. The plot shows that 20% of the molecular fragment or carbon atoms are left as residue beyond 450 °C. The degradation plot of temperature vs weight loss for compound (III) is shown in Fig. 5(III).
Unlike the three acridinium compounds, the acridine compound (IV) undergoes a single-stage degradation. At 450 °C, the compound starts degrading (or losing the weight percentage) expected with the breakdown of 1,8-dihydroxynaphthalene fragment and the weight loss continues up to 1000 °C. The compound degrades completely at 1000 °C leaving 20% of the molecular fragment or carbon atoms as residue beyond 1000 °C. The temperature vs weight loss plot for compound (IV) is shown in Fig. 5(IV). The thermal stability of compound (IV) compared to other acridinium compounds can be justified by the interaction energy calculation discussed in the above section. The calculated total energy value of intermolecular interaction (O‒H…N, O‒H…O, C‒H…O, C‒H…N) between the molecular pairs observed in compound (IV) is stronger compared to other three acridinium compounds (see Fig. 10).
Conclusion
In conclusion, it is understood that a significant relationship has been observed between the intermolecular in the crystal structure and the calculated interaction energies, photophysical property, and thermal property of acridinium/acridine compounds (I) to (IV). The crystal structure of compounds (I) to (IV) exhibits one- and two-dimensional supramolecular frameworks built through various types of intermolecular interactions. In the acridinium derivatives (I) to (III), the supramolecular framework is mainly stabilized by the formation of Cl-assisted hydrogen bonds and weak interactions such as N‒H…Cl, O‒H…Cl, C‒H…Cl, π…Cl, and Cl…Cl, respectively, whereas in acridine compound (IV), the supramolecular framework is due to the O‒H…N hydrogen bond and C‒H…N, C‒H…O, and C‒H…π interactions. The nature and geometry of π-contacts exhibited between the acridine moieties are influenced by the formation of other strong intermolecular interactions. The presence of various intermolecular interactions in the crystal structure and the percentage contribution in the molecular packing is understood from the Hirshfeld surface analysis and molecular electrostatic potential calculation. In addition to these, the strength/stability of the crystal is understood from the calculated interaction energy values exhibited between the molecular pairs in the crystal. In the above discussion, it is observed that the calculated total energy value of intermolecular interaction/contacts observed between the molecular pairs in the acridine compound (IV) is stronger than the energy value of intermolecular interaction/contacts of acridinium derivatives (I to III). This can be related to the experimental degradation temperature observed in the thermogravimetric analysis of compounds (I) to (IV). The total energy value of the intermolecular interaction associated with the Cl− (acceptor) atom shows a significantly stronger value. However, the calculated total energy value of π-contact (π…π, O…π, N…π) is stronger compared to other weak interactions (C‒H…O, C‒H…Cl, C‒H…π). In addition to these, the photophysical characterization of compounds (I) to (IV) has been carried out in this research. All compounds exhibit a characteristic broad absorption band in the range of 320‒420 nm attributed to the absorption behavior of the acridine moiety. The emission spectra show characteristic peaks between the range of 400‒500 nm. From the photophysical analysis, it is to be concluded that the acridinium derivatives (I to III) exhibit good photophysical behavior when compared with the acridine compound. The photoluminescence quantum yield for the acridinium derivatives (I) and (II) show a significant value when compared to the acridinium derivative (III), whereas the quantum yield for the acridine compound (IV) experiences a photoluminescence quenching resulting in very low quantum yield.
Data availability
Four crystal structures have been used in the manuscript. The data for the crystal structures is available in the Cambridge Crystallographic Data Centre in CIF format (Id CCDC2357373, 2357374, 2357375, and 2357376). FT-IR spectrum, interaction energy calculation output table is available in the supplementary information/supporting information. Other datasets analyzed during the current study are available from the corresponding author on reasonable request.
References
Lai CS, Anitha R, Lok YH, Chan SM, Harish R, Rohini K, Veerasamy R (2022) Struct Chem 33:649–669
Mélanie F, Natália T, Paula G, Cátia T (2021) Molecules 26:600
Dollinger S, Löber S, Klingenstein R, Korth C, Gmeiner P (2006) J Med Chem 49(22):6591–6595
Viana JO, Silva e Souza E, Sbaraini N, Vainstein MH, Gomes JNS, Olímpio de Moura R, Barbosa EG (2023) Sci Rep 13:7320
Kudryavtseva TN, Lamanov AY, Klimova LG, Nazarov GV (2017) Russ Chem Bull 66:123–128
Aarjane M, Slassi S, Tazi B, Maouloua M, Amine A (2020) Struct Chem 31:1523–1531
Varakumar P, Rajagopal K, Aparna B, Raman K, Byran G, Lima CMG, Rashid S, Nafady MH, Emran TB, Wybraniec S (2023) Molecules 28:193
Mróz A, Ryska I, Sominko H, Bejrowska A, Mazerska Z (2018) Pharmacol Rep 70:972–980
Elangovan A, Chiu HH, Yang SW, Ho TI (2004) Org Biomol Chem 2:3113–3118
Natrajan A, Sharpe D, Wen D (2012) Org Biomol Chem 10:3432–3447
Czechowska J, Kawecka A, Romanowska A, Marczak M, Wityk P, Krzymiński K, Zadykowicz B (2017) J Lumin 187:102–112
Nakazono M, Nanbu S, Akita T, Hamase K (2020) J Photochem Photobio A Chem 403:112851
Roda A, Guardigli M (2012) Anal Bioanal Chem 402:69–76
Prabakaran K, Manivannan R, Oh H, Parthiban C, So YA (2021) Dyes and Pig 192:109391
Du J, Li J, Li Y, Wang D, Cao H, He W, Zhou Y (2022) RSC Adv 12:3380–3385
Jagan R, Sivakumar K (2020) Struct Chem 31:917–925
Wera M, Storoniak P, Serdiuk IE, Zadykowicz B (2016) J Mol Struct 1105:41–53
Kandhan S, Krishnan P, Vansu E, Sahoo T, Jagan R, Srinivasan S, Aravindhan S, Gunasekaran S, Anbalagan G (2020) J Mater Sci 55:8591–8609
Song Y, Pan G, Zhang C, Wang C, Xu B, Tian W (2023) Mater Chem Front 7:5104–5119
Wang S, Shen Y, Zhang X, Liu H, Zhang ST, Li W, Yang B (2022) Dyes and Pig 205:110527
Ochi J, Tanaka K, Chujo Y (2023) Angew Chem Int Ed 62:e202214397
Zbačnik M, Kaitner B (2014) Cryst Eng Comm 16:4162–4168
Achar TK, Bose A, Mal P (2017) Beilstein J Org Chem 13:1907–1931
Avila-Ortiz CG, Juaristi E (2020) Molecules 25:3579
Banerjee M, Bhosle AA, Chatterjee A, Saha S (2021) J Org Chem 86:13911–13923
Bruker (2016) APEX3, SAINT-Plus and SADABS, Bruker AXS Inc., Madison, Wisconsin, USA
Sheldrick GM (2015) Acta Crystallogr C 71:3–8
Spek AL (2009) Acta Crystallogr D 65:148–155
Dolomanov OV, Bourhis LJ, Gildea RJ, Howard JAK, Puschmann H (2009) J Appl Crystallogr 42:339–341
Macrae CF, BrunoIJ, Chisholm JA, Edgington PR, McCabe P, Pidcock E, Rodriguez-Monge L, Taylor R, van de Streek J, Wood PA (2008) J Appl Crystallogr 41:466–470
Hwang J, Li P, Smith MD, Warden CE, Sirianni DA, Vik EC, Maier JM, Yehl CJ, Sherrill CD, Shimizu KD (2018) J Am Chem Soc 140:13301–13307
Spackman PR, Turner MJ, McKinnon JJ, Wolff SK, Grimwood DJ, Jayatilaka D, Spackman MA (2021) J Appl Crystallogr 54:1006–1011
Spackman MA, Jayatilaka D (2009) Cryst Eng Comm 11:19–32
Turner MJ, Grabowsky S, Jayatilaka D, Spackman MA (2014) J Phys Chem Lett 5:4249–4255
Mackenzie CF, Spackman PR, Jayatilaka D, Spackman MA (2017) IUCrJ 4:575–587
Pedzinski T, Marciniak B, Hug GL (2002) J Photochem Photobio A Chem 150:21–30
Kumar V, Singh DK, Mohan S, Hasan SH (2016) J Photochem Photobio B Bio 155:39–50
Datta S, Samanta S, Chaudhuri D (2018) J Mater Chem A 6:2922–2926
Schael F, Rubin MB, Speiser S (1998) J Photochem Photobio A Chem 115:99–108
Chen K, Zhao J, Li X, Gurzadyan GG (2019) J Phys Chem A 123(13):2503–2516
Khayyat SA, Abaker M, Umar A, Alkattan MO, Alharbi ND, Baskoutas S (2012) J Nanosci Nanotech 12(11):8453–8458
Acknowledgements
The authors thank the X-Ray Diffraction Facility (NSF-MRI, CHE-1827756), Department of Chemistry and Biochemistry, University of Missouri, St. Louis, for single crystal intensity data collection of compounds. The authors thank Dr. Keith Stine, Chair, Department of Chemistry and Biochemistry, University of Missouri, St. Louis, for helping with the procurement of chemicals.
Author information
Authors and Affiliations
Contributions
Jagan Rajamoni contributed to synthesis, crystal structure determination, computational studies, and manuscript preparation. Saptarshi Datta contributed to the analysis of FT-IR, TGA, UV-Vis, and photoluminescence studies. Jinjia Xu contributed to the discussion on photophysical characterization.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Rajamoni, J., Datta, S. & Xu, J. Crystal structure, supramolecular framework, hirshfeld surface analysis, and photophysical studies of some acridinium/acridine derivatives prepared by mechanochemical synthesis. Struct Chem (2024). https://doi.org/10.1007/s11224-024-02355-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11224-024-02355-9