
Abstract
A supramolecular cocrystal of 2-ammino-5-nitropyridine (ANP) with 2,4-dinitrophenol (DNP) has been successfully grown by the slow evaporation solution growth technique. The crystal structure of 2-amino-5-nitropyridine-2,4-dinitrophenol (ANDP) was elucidated by the single-crystal X-ray diffraction analysis, and it belongs to a monoclinic system with centrosymmetric space group P21/n. The bulk phase purity and homogeneity of the material were confirmed by powder X-ray diffraction analysis. The functional groups present in the molecule are identified by FT-IR analysis, and the band gap energy was estimated using diffuse reflectance data by the application of the Kubelka–Munk algorithm. The thermal stability of the compound was investigated by carrying out TG–DTA analysis. Theoretical calculations were performed using the density functional theory method to derive the dipole moment and hyperpolarizability. The high value of first-order molecular hyperpolarizability (β) suggests that it is a potential microlevel NLO candidate.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Supramolecular crystals have attractive molecular architectures for useful applications. Understanding intermolecular interactions can provide valuable information useful in the design of new crystals [1]. One or more delocalized bonds that, when exposed to electromagnetic fields, produce a high induced dipole moment, often in the form of a ring structure like benzene with donors and acceptors substituted at the para positions [2]. Modern research has shown that organic materials offer superior nonlinear optical (NLO) characteristics [3], optoelectronic materials [4], and data storage devices [5]. Phenol derivatives are interesting crystals, as the existence of the phenolic (–OH) group favors the formation of salts with several organic bases [6,7,8]. Additionally, by interactions with diverse organic compounds via π-bonding and hydrogen bonding, it forms cocrystals [9,10,11]. To some extent, molecular designs have been successful in improving charge transfer and first-order molecular hyperpolarizability (β). Intermolecular hydrogen bonding leads to the close packing of molecules within the crystal medium that is necessary for efficient nonlinear optical properties, along with van der Waals, molecular dipolar, and quadruple forces for energy minimization [12]. Many organic compounds with photosensitivity and thermal stability have recently been developed that are helpful in creating different crystalline electro-optical systems [13, 14].
Herewith, we described the synthesis, crystal structure, and supramolecular interactions of the 2-amino-5-nitropyridine-2,4-dinitrophenol (ANDP). It was analyzed by single-crystal and powder X-ray diffraction, UV diffuse reflectance, and photoluminescence spectroscopy and thermal analysis. Furthermore, it was investigated by Hirshfeld surface analysis, and theoretical studies are also presented in detail.
Experimental
Synthesis
2-Ammino-5-nitropyridine (ANP) and 2,4-dinitrophenol (DNP) were purchased from Alfa Aesar (assay: 99%) and commercially available solvents (Merck) were used without further purification. The cocrystal was synthesized by mixing equimolar quantities of 2-ammino-5-nitropyridine and 2,4-dinitrophenol in ethanol. The mixture was stirred at room temperature for 4 h (Scheme 1). The product was purified by recrystallization using ethanol as solvent. The transparent crystals were harvested in 7–10 days.

Synthesis of ANDP
Characterization techniques
A Bruker D8 QUEST single-crystal X-ray diffractometer (MoKα radiation (λ = 0.71073 Å) was used for single-crystal XRD studies. The Bruker APEX3 program was used for computing data collection, APEX3/SAINT using for the cell refinement, SAINT/XPREP was used for calculating the data reduction, SHELXT-2014/5 program was used for structure solution, SHELXL-2014/7 program was used for the structure refinement process and ORTEP3 program was used for molecular graphics. A Philips Xpert pro-triple-axis X-ray diffractometer (CuKα radiation in the 2θ range of 10–50°) was used for powder X-ray diffraction analysis. A Shimadzu IR Affinity-1 FT-IR spectrophotometer was used for the recorded FT-IR spectrum. The ultraviolet diffuse reflectance spectrum (UV–DRS) was recorded by the Shimadzu UV-2600 spectrophotometer. The photoluminescence spectrum was recorded by HJY:Fluorolg F3-111 fluorescence spectrometer. Hirshfeld surfaces and fingerprint plots were generated by CrystalExplorer 3.1 program [15]. All theoretical calculations were performed using the GAUSSIAN09W program using density functional theory (DFT) with B3LYP method 6-311G(d,p) as the basis set [16].
Results and discussion
XRD analysis
The as-grown crystal was finely powdered and subjected to powder XRD analysis. The bulk phase purity and homogeneity of ANDP were confirmed by matching experimental diffractograms with calculated patterns from the single-crystal XRD data. No significant variations are observed in peak positions of experimental and simulated XRD patterns (Fig. 1).

Simulated and experimental powder XRD patterns
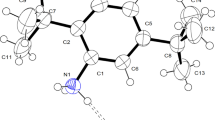
The crystal structure was determined by single-crystal X-ray diffraction and the crystallographic data are shown in Table 1. Selected bond lengths, angles, and torsion angles are listed in Table S1. Cocrystal ANDP grown from ethanol crystallizes in the centrosymmetric monoclinic system with space group P21/n and Z = 4. A displacement ellipsoid plot and molecular packing diagram are shown in Fig. 2. ANP and DNP molecules alternate in zig-zag chains, and the hydrogen atom was not transferred from the DNP to ANP due to the phenolic H atom was strongly interact with the intramolecular hydrogen bonding interactions, supramolecularly assembled by N–H•••O, N–H•••N, O–H•••O, O–H•••N, and C-H•••O hydrogen bonds. An infinite column-like structure is formed via π•••π staking interactions between ANP and DNP as shown in Fig. 3a, and it matches the structural arrangement of paracetamol with picric acid [11]. The centroid–centroid contact distance range is 3.609 to 3.896 Å with an angle ranging from 19.13 to 22.59°. Plane-to-plane distance ranges from 3.427 to 3.596 Å. Figure 3b displays the N–H•••O and N–H•••N intermolecular interactions. The intramolecular interaction (O(5)-H(5A)•••O(6)) is the shortest hydrogen bond observed in the DNP molecule with a distance of 2.6047(18) Å (Fig. 4a and b) shows the weak C-H•••O interactions and they are listed in Table 2.

a ORTEP and b molecular packing diagram of ANDP

The structural details: a π•••π interactions and b molecular packing interactions of N–H•••O and N–H•••N

Molecular packing interactions along a-axis: a O–H•••O and b C-H•••O interactions of ANDP
FT-IR spectroscopy
The vibrational spectrum of ANDP exhibits the characteristic bands of 2-amino-5-nitropyridine (ANP) and 2,4-dinitrophenol (DNP). The IR spectra of 2-amino-5-nitropyridine (SDBS No: 4210), 2,4-dinitrophenol (SDBS No: 2390) and ANDP are shown in Fig. S1. The band observed at 3295 and 3272 cm−1 corresponds to the symmetric stretching vibration of the phenolic O–H group of ANDP and DNP respectively. The absorption band around 3457 and 3463 cm−1 are attributed to the N–H stretching frequency of the amino group of ANDP and ANP respectively. The N–H bending vibration is observed at 1627 and 1637 cm–1 for ANDP and ANP respectively. The − NO2 group asymmetric and symmetric stretching appeared at 1568 and 1325 cm−1 for ANDP, 1537 and 1334 cm−1 for DNP and 1572 and 1336 cm−1 for ANP. The aromatic C-H stretching frequencies appear around 3093 cm−1 for ANDP, 3069 cm−1 for ANP, and 3078 cm−1 for DNP. The aromatic ring C = C stretching vibrations are assigned to peak at 1595 cm−1.
Optical studies
The optical reflectance spectrum of ANDP (Fig. 5) reveals high transmittance and the cut-off wavelength is ~ 434 nm. The band gap is estimated using the Tauc plot, [F(R)hʋ]2 versus hʋ and the estimated direct band gap energy of ANDP is 2.79 eV. The cocrystal exhibits fluorescent emission at λmax 649 nm (~ 1.91 eV) under the excitation at 434 nm in the solid state at room temperature (Fig. 6).

UV–Vis spectrum of ANDP (Tauc plot is given as an inset)

Photoluminescence spectrum of ANDP
Thermal analysis
The thermal stability of the compound was determined by thermogravimetric (TG) and differential thermal analysis (DTA). The TG–DTA response curve is presented in Fig. 7. In the DTA curve, the endothermic peak observed at 112 °C is attributed to the melting of the cocrystal, whereas the melting point of the reactants are 108 and 186 °C for DNP [17] and ANP [18] respectively. The TG curve specifies the major weight loss observed from 185 to 308 °C (mass loss 82.5%) indicating the decomposition of the sample. No exothermic or endothermic peak was detected below the melting point endotherm, representing the absence of any isomorphic phase transition in the specimen [19].

TG–DTA Curve of ANDP
Hirshfeld surface analysis
Figure 8 shows surfaces that have been mapped over dnorm, di, de, shape index, and curvedness. The surfaces are shown as transparent to allow visualization of the molecule around which they were calculated. Hydrogen bonding contacts are revealed by circular depressions (deep red) observable on the Hirshfeld surface, and the other visible spots in Fig. 8 are due to O•••H;H•••O (42.1%) interactions.

Hirshfeld surface: a dnorm, b di, c de, d shape index, and e curvedness
The combination of de and di in the form of a two-dimensional fingerprint plot provides a summary of intermolecular contacts in the crystal (Fig. 9). The O•••H:H•••O (42.1%) interactions are represented by a spike at the inner bottom of the plot in the inner top left region. The H•••H (12.2%) interactions are observed at the center of the fingerprint plot. In the top right corner, O•••O (9.3%) interactions are shown. Strong interactions occupy more space and weak interactions occupy less space in the fingerprint plot. Quantification of all intra- and intermolecular interactions are displayed as a pie chart Fig. S2.

Fingerprint plots
First-order molecular hyperpolarizability
The NLO responses in organic compounds originate from the π-electron excitation of individual molecules. The molecular level NLO response is confirmed by computational studies. Calculated polarizability (α), first-order molecular hyperpolarizability (β), and dipole moment (μ) of the specimen are 27.6680 × 10−24 esu, 1.6596 × 10−30 esu (∼9 times of urea), and 5.8347 D, respectively (Table 3). As shown in the table, βxxy tensor mainly contributes to the nonzero β value. Generally, a high βtot value is ascribed to conjugation, molecular symmetry, hyperconjugation, substitution, aromaticity, and charge transfer [20]. The high value of β suggests that the title compound has a good microlevel NLO response.
Frontier molecular orbital (FMO) analysis
Figure 10 displays the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), calculated using B3LYP/ 6-311G(d,p) as basis set and 2D images of orbitals computed in the gaseous state. The red color indicates the positive value and the green color indicates the negative value of the wave function. The HOMO is localized on the ANP molecule, whereas LUMO is localized on the DNP molecule. Both HOMO and LUMO are mostly π-antibonding type orbitals. Orbital is plotted at an isosurface value of 0.02 a.u. The calculated energy values are − 7.0352 eV (HOMO), − 4.0159 eV (LUMO), and 3.02 eV (energy gap). ΔEHOMO-LUMO explains the eventual charge transfer interactions within the molecule. A small frontier orbital gap facilitates polarizability and hence the title cocrystal is a promising candidate for NLO applications. The band gap derived from FMOs is higher than the experimental value. The experimental band gap is 2.79 eV, whereas the theoretical band gap is 3.02 eV. Variation is justified since the theoretical studies are for the gas phase, whereas the experiments are done in the condensed phase [21].

Frontier molecular orbital
Mulliken population analysis
Mulliken charges arise from the Mulliken population analysis and provide a means of estimating partial atomic charges from calculations carried out by the methods of computational chemistry. Mulliken atomic charge distribution is displayed in Fig. S3. All the oxygen, carbon (C2, C6, C8, and C24) and nitrogen (N26 and N27) are negative charges behaving as electron donors, whereas the remaining carbons (C1, C4, C5, C18, C19, C21, and C22), all the hydrogen atoms and nitrogen (N10, N11, and N28) are acceptors revealing positive charge (Table 4).
Molecular electrostatic potential (MEP)
The MEP plot of electrostatic potential mapped on the constant electron density surface displaying electrostatic potential distribution is shown in Fig. S4. The most negative electrostatic potential surfaces are represented as the red color that is expected to be sites of protonation and nucleophilic attack, whereas most positive electrostatic potential specifies the blue color and the green denotes regions of zero potential. The color code of this map is in the range between − 5.729e−2 to + 5.729e−2. The negative region is mainly localized from nitro group oxygen atoms, whereas the positive region lies in the amino group H (− NH2) atoms of the ANP molecule and aromatic C-H of the DNP molecule (Fig. S4). The circular red spots indicate the short hydrogen bonding contacts are visualized by Hirshfeld surface maps (Fig. 8).
Conclusions
Single crystals 2-amino-5-nitropyridine-2,4-dinitrophenol were successfully grown and characterized by spectroscopic and single-crystal X-ray diffraction techniques. The crystallographic data indicate that the title compound crystallizes in the monoclinic system with centrosymmetric space group P21/n. Minimum absorbance in the visible region coupled with high first-order molecular hyperpolarizability (β) in comparison with standard urea reveals good microlevel NLO response of the title compound. Prominent O•••H/H•••O interactions contribute to microlevel nonlinearity. Substitution of suitable donor–acceptor groups in the aromatic ring could make it SHG-active. Molecular electrostatic potential surfaces visualize the relative polarity and population analysis suggests the possibility of inter- and intramolecular charge transfer interactions responsible for observed nonlinearity.
Abbreviations
- ANDP:
-
2-Amino-5-nitropyridine-2,4-dinitrophenol
- ANP:
-
2-Ammino-5-nitropyridine
- DNP:
-
2,4-Dinitrophenol
- FT-IR :
-
Fourier transform infrared spectroscopy
- TG-DTA:
-
Thermogravimetric and differential thermal analysis
- NLO:
-
Nonlinear optical
- SHG:
-
Second harmonic generation
- MEP :
-
Molecular electrostatic potential
- DFT :
-
Density functional theory
- FMO :
-
Frontier molecular orbitals
- HOMO :
-
Highest occupied molecular orbital
- LUMO :
-
Lowest unoccupied molecular orbital
- UV–Vis DRS:
-
Ultraviolet visible near-infrared diffuse reflectance spectroscopy
- XRD:
-
X-ray diffraction
- CCDC :
-
Cambridge Crystallographic Data Centre
References
Wang K, Duan D, Wang R, Lin A, Cui Q, Liu B, Cui T, Zou B, Zhang X, Hu J, Zou G (2009) Stability of hydrogen-bonded supramolecular architecture under high pressure conditions: pressure-induced amorphization in melamine−boric acid adduct. Langmuir 25(8):4787–4791
Ng SL, Patil PS, Razak IA, Fun HK, Dharmaprakash SM (2006) A cocrystal of 1-(4-methoxyphenyl)-3-(3, 4, 5-trimethoxyphenyl) prop-2-en-1-one and (E)-3-(3-chloro-4, 5-dimethoxyphenyl)-1-(4-methoxyphenyl)-2-propen-1-one (0.92/0.08). Acta Cryst E 62(4):o1228-o1230
Basu S (1984) A review of nonlinear optical organic materials. Ind Eng Chem Prod Res Dev 23(2):183–186
Liu C, Bard AJ (1999) Optoelectronic properties and memories based on organic single-crystal thin films. Acc Chem Res 32(3):235–245
Ostroverkhova O (2016) Organic optoelectronic materials: mechanisms and applications. Chem Rev 116:13279–13412
Jin ZM, Pan YJ, Xu DJ, Xu YZ (2000) The 1: 1 complex of 4-nitrophenol and 4-methylpyridine. Acta Crystallogr C 56(2):e69–e70
Indumathi C, Girisun TS, Anitha K, Raj SAC (2017) Influence of number and position of nitro groups in tuning the thermodynamic and nonlinear optical properties of ethylenediaminium nitrophenolates. J Mol Liq 238:89–95
Manonmani M, Balakrishnan C, Meenakshisundaram SP, Sockalingam RM (2019) Synthesis, structural characterization and Hirshfeld surface analysis of 4-hydroxyanilinium picrate hydrate. Mater Today 15:645–653
Karuppasamy P, Kamalesh T, Anitha K, Kalam SA, Pandian MS, Ramasamy P, Verma S, Rao SV (2018) Synthesis, crystal growth, structure and characterization of a novel third order nonlinear optical organic single crystal: 2-amino 4, 6-dimethyl pyrimidine 4-nitrophenol. Opt Mater 84:475–489
Moreno-Fuquen R, Cano F, Martinez-Ripoll M, Montano A, Zukerman-Schpector J (2001) 1:1 Complex of 2,4-dinitrophenol and 4-methoxypyridine N-oxide hydrate. Acta Crystallogr E 57:o712–o714
Manonmani M, Balakrishnan C, Ahamed SR, Vinitha G, Meenakshisundaram SP, Sockalingam RM (2019) Cocrystallization of paracetamol-picric acid: Hirshfeld surface analysis, supramolecular architecture and third-order nonlinear optical properties. J Mol Struct 1190:1–10
Reshak AH, Alahmed ZA, Azam S (2014) Electronic structure, electronic charge density and optical properties analyses of Rb2Al2B2O7 compound: DFT calculation. Int J Electrochem Sci 9:975–989
Williams D (1983) Nonlinear optical properties of organic and polymeric materials. American Chemical Society, Washington, DC
Phrutivorapongkul A, Lipipun V, Ruangrungsi N, Kirtikara K, Nishikawa K, Maruyama S, Watanabe T, Ishikawa T (2003) Studies on the chemical constituents of stem bark of Millettia leucantha: isolation of new chalcones with cytotoxic, anti-herpes simplex virus and anti-inflammatory activities. Chem Pharm Bull 51:187–190
Wolff SK, Grimwood DJ, McKinnon JJ, Turner MJ, Jayatilaka D, Spackman MA (2012) CrystalExplorer (Version 3.1). University of Western Australia
Frisch MJ et al (2009) GAUSSIAN09, Revision C.01. Gaussian, Inc., Wallingford, CT, USA
https://www.chemicalbook.com/ProductList_en.aspx?kwd=2,4-Dinitrophenol
https://www.chemicalbook.com/ProductList_en.aspx?kwd=2-amino-5-nitropyridine
Manikandan A, Manivannan D, Dhanalakshmi M, Guganathan L, Kokila T, Santhamoorthy M, Markkandan R, Kim SC, Balakrishnan C (2022) Synthesis, structural characterization and Hirshfeld surface analysis of NLO active 3-hydroxypyridinium 2,4-dinitrophenolate single crystals. J Mater Sci Mater Electron 33:7750–7764
Zyss J, Oudar JL (1982) Relations between microscopic and macroscopic lowest-order optical nonlinearities of molecular crystals with one- or two-dimensional units. Phy Rew A 26(4):2028
Jauhar RM, Murugakoothan P (2017) Optimization, frontier molecular orbitals and dielectric studies on 2,6-diaminopyridiniumTosylate single crystals. AIP Conf Proc 1832:100005–100011
Funding
The authors thank the Council of Scientific and Industrial Research, New Delhi, for financial support through research grant No. 21 (1024)/16/EMR-II. SS and CB are grateful to the CSIR Emeritus Scientist Scheme for the award of the SRF/RA-ship.
Author information
Authors and Affiliations
Contributions
S S: data curation, investigation, visualization, validation, writing, original draft. C B: data curation, investigation, visualization, validation. P S: investigation. SP M: review, editing, and supervision. The final version of the manuscript submitted was approved by all the authors.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sivaraman, S., Balakrishnan, C., Suppuraj, P. et al. Synthesis, structure, Hirshfeld surface analysis, and computational studies of 2-amino-5-nitropyridine-2,4-dinitrophenol cocrystal. Struct Chem 34, 239–249 (2023). https://doi.org/10.1007/s11224-022-02087-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-022-02087-8