
Abstract
1,2,4-Triazole is a popular scaffold in drug design. According to chemical nature, the triazole ring tends to prototropic tautomerism. Tautomeric phenomena are important for studying the chemical reactivity and interaction of drugs based on triazole with biomolecules in the human body. Theoretical modeling was used to assign structures of newly synthesized 2-(3-hetaryl-1,2,4-triazol-5-yl)anilines. The procedure included quantum-chemical SMD/M06-2X/6–311++G(d,p) calculation of the relative stability for possible tautomers, simulation of UV/vis spectra for the most stable forms, and comparison of the resulting curves with the experimental spectral data taking into account the Boltzmann weighting. The influence of the substituents in triazole ring on tautomeric equilibrium was elucidated. NBO charge distribution, dipole moment, molecular electrostatic potential, and HOMO/LUMO gap for the most stable tautomers were analyzed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The chemistry of 1,2,4-triazoles has received considerable attention in recent years due to their usefulness in different areas of biological activities and as industrial intermediates. Several reviews have addressed the therapeutic potential of 1,2,4-triazole derivatives, which possess antimicrobial, antimalarial, antitubercular, antiviral, antioxidant, anti-inflammatory, anticonvulsant, antipyretic, vasoconstriction, and anticancer activities [1,2,3,4,5,6,7,8]. Triazole moiety is used as significant components in new synthetic azo dyes to enhance their color strength and brilliant shades [9] and in metal complexes with the ability of photo- and electroluminescence [10]. Substituted 1,2,4-triazoles have found application in agriculture and industries as plant growth regulators [11], metal corrosion inhibitors [12], and energetic materials [13].
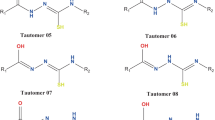
Triazole ring exhibits different tautomeric forms. There are three possible tautomers of C-substituted 1,2,4-triazoles depending on the position of hydrogen atom in the ring (Scheme 1). Tautomeric preferences and factors affecting the equilibrium are important to understand the chemical reactivity of compounds and their impact on biological systems. NMR, IR, UV/vis, microwave, and mass spectra and X-ray diffraction are used to experimentally determine the most abundant tautomer in gas, solid, and dissolved states. Theoretical computer modeling may predict the most stable tautomeric forms and their relative free energy, calculate population, and explain the tautomeric equilibrium by analysis of molecular electronic properties. Combination of experimental and theoretical methods is a powerful tool to study the tautomeric phenomena.

Structure of 1,2,4-triazoles with atom numbering
Experimental studies confirmed the existence of unsubstituted 1,2,4-triazole in N1-H form in the vapor phase by microwave spectroscopy [14, 15], in the crystalline state by neutron diffraction [16], and in DMSO solution by 15 N NMR [17]. Computational studies of 1,2,4-triazole tautomerism showed that the N1-H form is more stable than the N4-H tautomer by more than 6 kcal/mol [18,19,20,21,22]. The high stability of tautomer N1-H may be attributed to the reduced repulsion of the unshared electron pairs of the nitrogen atoms in these species in comparison with N4-H, that was seen in the computed electrostatic potential distribution [23].
The various substituents present in the triazole ring affect the stability of the tautomeric forms in different ways. X-ray and NMR studies confirm that 3-halo-1,2,4-triazoles, 3-(2-hydroxyethylsulfanyl)-5-phenyl-1,2,4-triazole, and 3-(2-hydroxyethylsulfanyl)-5-methyl-1,2,4-triazole exist as N1-H tautomers in solid phase and solution [24, 25]. The N1-H form of 3-nitro-1,2,4-triazole is the most stable in dioxane and chloroform that was confirmed by measured dipole moment [26]. IR spectroscopy showed a mixture of N1-H and N2-H forms for 5-(1H-tetrazol-1-yl)-1,2,4-triazole [27], and 3-amino-1,2,4-triazole, with domination of the first tautomer [28]. Experimental and computational studies indicate that N2-H is the most stable tautomer for 3-amino-5-nitro-1,2,4-triazole and 3-amino-1,2,4-triazole in crystal and polar solvent [29,30,31]. NMR and UV spectra indicate that a tautomeric hydrogen atom of the predominant tautomer of 3,5-disubstituted 1,2,4-triazoles is attached to the nitrogen atom at position 1 or 2 which is closer to the more electron-releasing substituents in the 3 or 5 position [32].
The tautomerism of 3-amino-5-(het)aryl-1,2,4-triazoles was investigated using NMR spectroscopy and X-ray crystallography. The triazoles were found to exist in N1-H and N2-H forms, the N4-H form was not observed either in the solid state or in solution [33]. The thermodynamic stability of N2-H form in comparison with N1-H increased together with the electron-withdrawing properties of the substituents. In case of heterocyclic substitutes, the intramolecular hydrogen bonding N1-H…X (X = O, N) would prevent shifting of the tautomeric equilibrium towards “electronically” favored form N2-H despite the strong electron-withdrawing effect of the heterocycles.
DFT studies on tautomerism of C5-substituted 1,2,4-triazoles showed that the N4–H form is the least stable tautomer, which is less stable than N2-H or N1-H form by more than 5 kcal/mol [34]. The electron-donating substituents (–OH, –F, –CN, –NH2, and –Cl) stabilize the N2–H tautomer, whereas the electron-withdrawing substituents (–CONH2, –COOH, –CHO, –BH2, and –CFO) stabilize the N1–H tautomer of C5-substituted 1,2,4-triazoles. Experimental UV/vis spectra and theoretical modeling of isomeric 2-(3-(methoxyphenyl)-1,2,4-triazole-5-yl)anilines showed that N2-H tautomer dominates for o-methoxy isomer, while the molecules of m- and p-methoxy isomers exist as a mixture of N1-H and N2-H tautomeric forms [35]. Summarizing the abovementioned experimental and theoretical results, one can see that N4-H tautomer is the least abundant form for 1,2,4-triazoles, and the preference of N1-H or N2-H form depends on substituents and environment.
The aim of the present study is a theoretical investigation of tautomerism for synthesized 2-(3-hetaryl-1,2,4-triazol-5-yl)anilines. The task is important to predict reactivity, including regiochemistry, for these compounds and to further study their potential biological activity.
Experimental and computational methods
All calculations were carried out using the Gaussian 09 suite of programs [36]. The geometry optimization was performed at M06-2X level in conjunction with Pople’s type 6–311+ +G(d,p) basis set. To account for the solvent effect, the SMD model with methanol (ε = 32.613) as a solvent was applied. The UV–vis calculations were performed at SMD/PBE1PBE level using developed in our group STO##-3Gel basis set [37], which demonstrated high efficiency for electric and magnetic properties calculation [35]. The intensities of the calculated spectra were scaled using Gabedit program [38] to fit the most intensive band obtained experimentally. The NBO atomic charges, frontier molecular orbitals properties, and dipole moments are calculated at SMD/M06-2X/6–311++G(d,p) level.
The ultraviolet–visible spectra of compounds 1–6 solved in methanol were recorded by using the double-beam UV–vis spectrophotometer SPECORD 200 in the region of 200–380 nm at room temperature.
Results and discussion
Conformational analysis and population of tautomers
Six synthesized 2-(3-hetaryl-1,2,4-triazol-5-yl)anilines 1–6 were used to study their tautomeric behavior [39]. The correspondent 3-hetaryls are benzofuran-2-yl (1), benzothiophen-2-yl (2), indol-2-yl (3), furan-2-yl (4), thiophen-2-yl (5), and pyrrol-2-yl (6). Each of the compounds has three tautomers A, B, and C according to the position of hydrogen atom in the triazole ring N1-H, N2-H, and N4-H, respectively (Fig. 1). Each tautomer has four conformers (c1–c4) due to the ability to rotate substitutes in positions 3 and 5 (Fig. 1). That is, for 72 structures, free Gibbs energy and population were calculated. The results obtained in the methanol and gas phase are shown in Tables 1 and S1, respectively. Optimization of the geometry for different torsion angles ψ1 and ψ2 made it possible to localize all possible conformations (Fig. 1). For an investigation of substitute influence on the stability of different tautomers, the unsubstituted in position 3 of the triazole ring compound 2-(1,2,4-triazol-5-yl)aniline (7) was modeled. Its calculated characteristics are provided in Tables S2 and S5.

Tautomers A–C and conformers c1–c4 for triazoles 1–7
The most representative tautomers for compounds 1–7 are A and B both in the gas phase and in methanol (Tables 1, S1 and S2). Structures of the 2-(3-hetaryl-1,2,4-triazol-5-yl)anilines tautomeric forms, that have population more than 5%, are represented in Fig. 2. They attend to have structure near to planar (Tables 1 and S1). The structures 1B-c1, 2B-c3, 3B-c3, 4B-c1, 4B-c3, 5B-c3, 6B-c3 are highly planar, which promotes aromaticity and thus raises their population. Several intramolecular hydrogen bonds provide stabilization effect (Figs. 2 and S1). Hydrogen bonds in tautomer A are formed between the hydrogen of the amino group and nitrogen N5 of triazole, and between the hydrogen of hetaryl substitute and nitrogens N2 and N5 of triazole. In tautomer B, hydrogen bonds may be formed by hydrogen of amino group and hydrogen of phenyl with nitrogens N5 and N1 of triazole, between hydrogen of hetaryl substitute and nitrogen N5 of triazole, and between hydrogen of N2-H of triazole and heteroatom of hetaryl substitute. Tautomer C has the highest relative energy due to steric distortion (Tables 1 and S1) and destabilizing vicinity of two lone electron pairs at the N1 and N2 atoms, that was also observed earlier for 5-substituted 1,2,4-triazoles [34].

SMD/M06-2X/6–311++G(d,p) calculated selected intramolecular hydrogen bonds for the most stable tautomer of triazoles 1–6 in methanol
Gas phase favors for tautomer A of compounds 2, 3, 5, and 7. Furane and benzofurane at position 3 of 1,2,4-triazole considerably increase stability of B tautomer in gas phase as compared with 3-unsubstituted 1,2,4-triazole (7) (Tables S1 and S2), which may be explained by the formation of additional stronger intramolecular hydrogen bonds between atoms of hetaryl moiety and triazole ring for B tautomer than for A tautomer (Fig. S1). Geometry of compounds 1–7 slightly changes in methanolic solution as compared with gas phase (Figs. 2, S1 and S2). The hydrogen bond distance, mostly, slightly increased in solution. Methanol significantly stabilizes tautomer A for compounds 1 and 4, which may be due to the high electronegativity of oxygen of hetaryl moiety and its ability to form intermolecular hydrogen bonds with methanol molecules to increase the stability of the tautomer A (Fig. 2). While oxygen atom of tautomer B of compounds 1 and 4 is involved in the formation of intramolecular bond with hydrogen of aminogroup. Tautomer A is a prevalent form in methanol for compounds 1–5, with the order of population as 1 > 3 > 4 > 2 > 5.
Theoretical modeling of isomeric 2-(3-(methoxyphenyl)-1,2,4-triazole-5-yl)anilines, which are close to structures of studied compounds, showed that N2-H (B) tautomer dominates for o-methoxy isomer because of formation of the intramolecular hydrogen bond H⋅⋅⋅O between hydrogen of N2-H and oxygen of methoxy group [35]. A mixture of tautomeric forms N1-H (A) and N2-H (B) was predicted for m- and p-methoxy derivatives where the degree of conjugation played the decisive role [35]. Our calculations also predict tautomers A and B to be the most representative for 2-(3-hetaryl-1,2,4-triazol-5-yl)anilines that are provided by intramolecular bond formation and high degree of conjugation because of the structure planarity (Tables 1, S1 and S2).
UV/vis spectra
Calculated UV/vis spectra for the most stable tautomers of compounds 1–6 are depicted in Fig. S3. The main electronic transitions for tautomeric forms of 1–6 are listed in Table S3. Energy of selected molecular orbitals involved in transitions are collected in Table S4. If the tautomer is represented by several conformations populated more than 5% as well as the existing of the tautomeric mixture, the Boltzmann weighted sum spectrum was generated (Fig. 3). All calculated curves were compared with the experimental data. Data for calculated and experimental absorption wavelengths λmax are gathered in Table 2.

Experimental UV/vis spectrum in methanol and SMD/PBE1PBE/STO##-3Gel computed spectra and Boltzmann weighted sum spectra for compounds 1–6
Compound 1 has two bands in experimental UV/vis spectrum with maxima at 219 and 286 nm. Calculated spectra for the most stable tautomers of 1 perfectly match the first absorption maxima (Table 2). There are two calculated bands in the region of the wide experimental second band (Fig. S3). The most intensive maximum of tautomers A are red-shifted relative to the one of tautomer B. Different conformers for tautomer A or B match to each other. The most intense peaks for different tautomers consist of several excitations (Table S3). The more long-wave band can be assigned to n → π* transition, while others – to π → π* ones. HOMO–LUMO transition contributes to the long-wave maximum. The lower lying HOMOs and upper lying LUMOs are involved in transitions in case of short-wave band.
Experimental UV/vis spectrum of compound 2 contains two peaks at 230 and 293 nm (Fig. 3, Table 2). Absorption maxima of compound 2 are bathochromically shifted as compared with compound 1. Calculated spectrum of tautomers A shows two peaks in the area of the second experimental band, while tautomer B shows one long-wave band that good matches the experimental one. The first peaks of both tautomers good match each other and are slightly blue-shifted according to experimental data. The same feature is observed for the calculated first peak of compound 3. The wavelengths for experimental bands of compound 3 are 225 and 303 nm. In the area of the second experimental band, tautomer B has two peaks, while there are three absorption maxima in the spectrum of tautomer A. Comparison of experimental spectra of compounds 1, 2, and 3 with the corresponding Boltzmann weighted sum spectrum shows satisfactory results (Fig. 3).
UV/vis spectrum of compound 4 contains three experimental peaks at 225, 254, and 325 nm. Tautomer A has three calculated bands, while tautomer B has four bands, two of which correspond to the first experimental peak (Fig. S3, Table 2). Tautomer A better matches the experimental spectra than tautomer B, that corresponds to a tautomers’ stability. Experimental spectrum of compound 5 has two bands at 218 and 317 nm and one shoulder at 260 nm (Fig. 3, Table 2). Despite the experimental spectrum of 5 does not have a well-defined peak in the middle region, the calculated spectra for both tautomers have several peaks (Fig. S3, Table 2). The second absorption maxima in simulated spectra correspond to the position of the shoulder in experimental spectra. Calculated UV–vis spectra of tautomer A are red-shifted as compared with one of tautomer B. Three absorption maxima at 221, 263, and 319 nm are observed in the experimental spectrum of compound 6. Calculated spectra show mostly the same feature as observed for compound 5. Comparison of experimental spectra of compounds 4–6 with the corresponding Boltzmann weighted sum spectra shows that the first and the second absorption maxima excellent match experimental bands. Meanwhile, the third peak in the spectra of tautomer A better corresponds to experimental data than one of tautomer B (Fig. 3, Table 2). We suggest experimental spectra may be a superposition of the UV–vis spectra of individual tautomers. There is no correlation between the wavelength of the shortest band of UV–vis spectra of compounds 1–6 and the HOMO/LUMO energy gap (Tables 2 and 4), which is in consistence with involving several molecular orbitals in transition. The calculations, performed at SMD/PBE1PBE level using developed in our group STO##-3Gel basis set [37], demonstrate high efficiency for UV–vis spectra simulation and allow to determine the relative stability of different tautomers of 2-(3-hetaryl-1,2,4-triazol-5-yl)anilines as well as 2-(3-(methoxyphenyl)-1,2,4-triazole-5-yl)anilines that were shown in our previous work [35].
Dipole moments
Table 3 contains calculated dipole moments for all compounds in the gas phase and methanolic solution. Different tautomeric forms have various electronic structures and, correspondently, different dipole moments. The increasing dipole moment in solution as compared with the gas phase is observed for all tautomers that suggest an increase of solute–solvent interaction. The largest increase of dipole moment for tautomer A is observed for compounds 3 and 6, for tautomer B — in case of compounds 2 and 5, that correspond to absolute value of dipole moment. There is a good correlation between dipole moments in solution and gas phase for all tautomers with R2 = 0.96. As shown in Table 3 and Table S5, dipole moment of tautomers depends on substitutes in the triazole ring. Tautomer B has higher dipole moment than tautomer A for compounds 1, 2, 4, 5, and 7, while the reverse situation is observed for compound 3. The differences in dipole moment between tautomers A and B are in a range of 1–3 D. The differences in dipole moment between different conformers c1–c4 are smaller and do not exceed 1.5 D.
Molecular electrostatic potential
The molecular electrostatic potential (MEP) shows the molecular charge distributions in terms of the atomic charge distributions. MEP mapped on electron density surface (Fig. S4) shows that tautomers A and B have different charge distribution. The most negative electrostatic potential in tautomer A is located on atoms O, N2, and N4 for compounds 1 and 4, on atoms S, N2, and N4 for compounds 2 and 5, on atom N2 and thienyl or benzothienyl moiety for compounds 3 and 6. In case of tautomer B, the most negative electrostatic potential is concentrated on atom N4 and aminophenyl substituent for all compounds. The most positive electrostatic potential is located on the hydrogen atom of the triazole ring in both tautomers, and additionally on hydrogens of the amino-group for tautomer A. The localizations of the most negative electrostatic potential are sites for electrophilic attack, while high concentration of positive electrostatic potential facilitates attack of nucleophile.
NBO charge distribution
Calculated charge distribution in 1,2,4-triazole ring using NBO technique for the most stable tautomers of compounds 1–7 in methanol is shown in Tables S5 and S6. Introduction of substituent in position 3 leads only to decrease charge of C3 in 0.1 e. Nitrogen atom at position 4 (N4) carries the most negative net charge in 1,2,4-triazole ring, while the nitrogen atom of the amino-group at phenyl ring has the most negative charge in molecule and predicted to be effective for interaction with electrophiles. Nitrogen atoms N1 and N2 have close values of charges, which differ from the charge of N4 by 0.19–0.27 e. Tautomer A has 0.1 e larger positive charge at C5 atom than at C3 atom. The influence of substituent nature at position 3 on charge of all atoms of 1,2,4-triazole ring for compounds 1–6 is insignificant.
Frontier molecular orbitals
The highest occupied molecular orbital (HOMO) energy is associated with ionization potential and the lowest unoccupied molecular orbital (LUMO) energy — with electron affinity. The difference between HOMO and LUMO energies (energy gap) is a measure of chemical reactivity. HOMO and LUMO surfaces for compounds 1–6 are shown in Fig. S5.
The localization of HOMO electrons in compounds 1 and 2 is centered on aminophenyl and triazole moieties. The electron distributions in HOMO of compounds 3–6 are mostly localized over whole molecule, excluding atom N4 in compound 3, atoms N4 and O in 4, atom S in 5, and atoms N4 and N of pyrrole in 6. LUMO is located on triazole and 3-substituent parts of molecule for compounds 1 and 2, and on aminophenyl and triazole moieties for compound 6. Tautomers A and B have different localization of LUMO for compounds 3–5. LUMO is located mostly on aminophenyl and triazole moieties in compounds 3 and 4, and over whole molecule in compound 5 excluding atom N4 and amino-group in case of tautomer A. For tautomer B, the localization of LUMO is observed on triazole and 3-substituent parts of molecule of compounds 3–5.
Introduction of pyrrole or indole in position 3 of 5-(2-aminophenyl)-1,2,4-triazole leads to an increase in HOMO energy in 0.1–0.2 eV, while the rest substitutes have an insignificant influence on HOMO level (Table 4). All substitutes decrease LUMO energy. The highest impact was observed after introducing benzofuran and benzothiophene, which causes lowering LUMO level in 0.73 and 0.88 eV, respectively. As a result of HOMO and LUMO energy changes, the energy gap for compounds 1–6 is 0.1–0.9 eV lower than for compound 7. That means the introduction of substitutes in position 3 increases the chemical reactivity of triazoles. The influence of substitutes follows row benzothiophen-2-yl > benzofuran-2-yl > thiophen-2-yl > indol-2-yl > furan-2-yl > pyrrol-2-yl.
Tautomer B has lower values for LUMO and lower energy gap than tautomer A for compounds 1, 2, 3, and 5, while the opposite situation is observed for compound 6. Different conformers for each tautomer have close values of molecular orbital energy. The difference does not exceed 0.1 eV.
Compounds 1, 2, and 3 have smaller ΔE, than compounds 4, 5, and 6, respectively. The presence of benzene ring fused to heterocycle in substitute decreases energy gap and facilitates reactivity, mainly by decrease of LUMO values, while change of HOMO energy is insignificant (Table 4). Values of LUMO energy are in a range from −0.17 to −0.94 eV for tautomeric forms of compounds 1–6. While for the HOMO energy, the values from −6.90 to −7.07 are observed.
The order of the energy gap is as follows: 2 < 1 < 3 < 5 < 4 < 6. The ability to attach an electron decreases in the row 2 > 1 > 5 > 3 > 4 > 6. Benzene ring and sulfur atom in a substitute facilitate electron affinity by the ability of negative charge delocalization.
Conclusions
The tautomerism of 2-(3-hetaryl-1,2,4-triazole-5-yl)anilines was examined by using theoretical technique. The obtained data suggest that all studied compounds exist as a mixture of N1-H and N2-H tautomers. The preference of tautomeric form strongly depends on the substitute in 1,2,4-triazole ring and environment. The gas phase favors N1-H form in case of thiophenyl, benzothiophenyl, and indolyl substituents, while furyl, benzofuryl, and pyrrolyl-substituted 1,2,4-triazole exist preferable in N2-H form. In methanolic solution, N2-H tautomer dominates only for pyrrolyl-substituted compound. The UV/vis simulated Boltzmann weighted sum spectra for two tautomers well reproduce the experimental ones. Substituents strongly influence the level of LUMO, while the change of atomic charges is insufficient. Tautomers N1-H and N2-H have different electrostatic potential distributions and HOMO/LUMO energy gap and, correspondently, different reactivity.
Data availability
The data supporting the findings of this study are available within its supplementary material and from the corresponding author on reasonable request.
Code availability
Gaussian 09 package program.
References
Sharma V, Shrivastava B, Bhatia R, Bachwani M, Khandelwa R, Ameta J (2011) Exploring potential of 1,2,4-triazole: a brief review. Pharmacologyonline 1:1192–1222 https://pharmacologyonline.silae.it/files/newsletter/2011/vol1/104.sharma.pdf
Kharb R, Sharma PC, Yar MS (2011) Pharmacological significance of triazole scaffold. J Enzyme Inhib Med Chem 26:1–21 https://doi.org/10.3109/14756360903524304
Aly AA, Hassan AA, Makhlouf MM, Brase S (2020) Chemistry and biological activities of 1,2,4-triazolethiones – antiviral and anti-infective drugs. Molecules 25:3036 https://doi.org/10.3390/molecules25133036
Song M-X, Deng X-Q (2018) Recent developments on triazole nucleus in anticonvulsant compounds: a review. J Enzyme Inhib Med Chem 33:453–478 https://doi.org/10.1080/14756366.2017.1423068
Ram VJ, Sethi A, Nath M, Pratap R (2017) The chemistry of heterocycles: nomenclature and chemistry of tree-to-five membered heterocycles. Elsevier https://doi.org/10.1016/C2015-0-05990-1
Kaur P, Chawla A (2017) 1,2,4-Triazole: a review of pharmacological activities. Int Res J Pharm 8:10–29 https://doi.org/10.7897/2230-8407.087112
Maddila S, Pagadala R, Jonnalagadda S (2013) 1,2,4-Triazoles: a review of synthetic approaches and the biological activity. Lett Org Chem 10:693–714 https://doi.org/10.2174/157017861010131126115448
Zhou C-H, Wang Y (2012) Recent researches in triazole compounds as medicinal drugs. Curr Med Chem 19:239–280 https://doi.org/10.2174/092986712803414213
Khanmohammadi H, Erfantalab M (2012) New 1,2,4-triazole-based azo-azomethine dyes. Part I: Synthesis, characterization and spectroscopic studies. Spectrochim Acta 86:39–43 https://doi.org/10.1016/j.saa.2011.09.053
Gusev AN, Shulgin VF, Minaev BF, Baryshnikov GV, Minaeva VA, Baryshnikova AT, Kiskin MA, Eremenko IL (2017) Synthesis and luminescent properties of copper(I) complexes with 3-pyridin-2-yl-5-(4-R-phenyl)-1H-1,2,4-triazoles. Russ J Inorg Chem 62:423–430 https://doi.org/10.1134/S1070363211110193
Jin J-Y, Zhang L-X, Zhang A-J, Lei X-X, Zhu J-H (2007) Synthesis and biological activity of some novel derivatives of 4-amino-3-(D-galactopentitol-1-yl)-5-mercapto-1,2,4-triazole. Molecules 12:1596–1605 https://doi.org/10.3390/12081596
Sherif E-SM, Erasmus R, Comins J (2007) Effects of 3-amino-1,2,4-triazole on the inhibition of copper corrosion in acidic chloride solutions. J Colloid Interface Sci 311:144–151 https://doi.org/10.1016/j.jcis.2007.02.064
Lee K-Y, Chapman LB, Cobura MD (1987) 3-Nitro-1,2,4-triazol-5-one, a less sensitive explosive. J Energ Mater 5:27–33 https://doi.org/10.1080/07370658708012347
Bolton K, Brown RD, Burden FR, Mishra A (1971) The microwave spectrum and dipole moment of 1,2,4-triazole: identification of tautomer in vapour phase. Chem Commun 15:873 https://doi.org/10.1039/C29710000873
Bolton K, Brown RD, Burden FR, Mishra A (1975) The microwave spectrum and structure of 1,2,4-triazole. J Mol Struct 27:261–266 https://doi.org/10.1016/0022-2860(75)87034-7
Jeffrey GA, Ruble JR, Yates JH (1983) Neutron diffraction at 15 and 120 K and ab initio molecular-orbital studies of the molecular structure of 1,2,4-triazole. Acta Crystallogr B 39:388 https://doi.org/10.1107/S010876818300258X
Bojarska-Olejnik E, Stefaniak L, Witanowski M, Webb GA (1986) 15N NMR investigation of the tautomeric equilibria of some 1,2,4-triazoles and related compounds. Magn Reson Chem 24:911–914 https://doi.org/10.1002/mrc.1260241013
Mo O, de Paz JLG, Yanez M (1986) Protonation energies and tautomerism of azoles: basis set effects. J Phys Chem 90:5597–5604 https://doi.org/10.1021/j100280a024
Billes F, Endredi H, Keresztury G (2000) Vibrational spectroscopy of triazoles and tetrazole. J Mol Struct (Theochem) 530:183–200 https://doi.org/10.1016/S0166-1280(00)00340-7
Cox JR, Woodcock S, Hillier IH, Vincent MA (1990) Tautomerism of 1,2,3- and 1,2,4-triazole in the gas phase and in aqueous solution: A combined ab initio quantum mechanics and free energy perturbation study. J Phys Chem 94:5499–5501 https://doi.org/10.1021/j100377a016
Claramunt RM, Sanz D, Alkorta I, Elguero J, Foces-Foces C, Llamas-Saiz AL (2001) Ab initio study of azolides: energetics and spectroscopic properties. J Heterocycl Chem 38:443 https://doi.org/10.1002/jhet.5570380221
Balabin RM (2009) Tautomeric equilibrium and hydrogen shifts in tetrazole and triazoles: focal-point analysis and ab initio limit. J Chem Phys 131:154307 https://doi.org/10.1063/1.3249968
Davarski KA, Khalachev NK, Yankova RZ, Raikov S (1998) Quantum chemical study of the tautomerism, geometry, and electronic structure of 1,2,3- and 1,2,4-triazoles. Chem Heterocycl Compd 34:568–574 https://doi.org/10.1007/BF02290940
Claramunt RM, Lopez C, Garcia MA, Otero MD, Torres MR, Pinilla E, Alarcon SH, Alkorta I, Elguero J (2001) The structure of halogeno-1,2,4-triazoles in the solid state and in solution. New J Chem 25:1061–1068 https://doi.org/10.1039/B103405G
Buzykin BI, Mironova EV, Nabiullin VN, Azancheev NM, Avvakumova LV, Rizvanov IKh, Gubaidullin AT, Litvinov IA, Syakaev VV (2008) Tautomerism of aza cycles: II.1 Synthesis and structure of 5-substituted 3-(2-hydroxyethylsulfanyl)-1H-1,2,4-triazoles and their alts. Psreference of the 1H,4H–1,2,4-triazolium tautomers. Russ J Gen Chem 78:461–479 https://doi.org/10.1134/S1070363208030225
Pevzner MS, Fedorova EY, Shokhor IN, Bagal LI (1971) Heterocyclic nitro compounds. Chem Heterocycl Compd 7:252–254. https://doi.org/10.1007/BF00473101
Pagacz-Kostrzewa M, Reva ID, Bronisz R, Giuliano BM, Fausto R, Wierzejewska M (2011) Conformational behavior and tautomer selective photochemistry in low temperature matrices: The case of 5-(1H-tetrazol-1-yl)-1,2,4-triazole. J Phys Chem A 115:5693–5707 https://doi.org/10.1021/jp202925r
Pagacz-Kostrzewa M, Bil A, Wierzejewska M (2017) UV-induced proton transfer in 3-amino-1,2,4-triazole. J Photoch Photobio A 335:124–129 https://doi.org/10.1016/j.jphotochem.2016.11.023
Sorescu DC, Bennett CM, Thompson DL (1998) Theoretical studies of the structure, tautomerism, and vibrational spectra of 3-amino-5-nitro-1,2,4-triazole. J Phys Chem A 102:10348–10357 https://doi.org/10.1021/jp9824712
Garcia E, Lee KY (1992) Structure of 3-amino-5-nitro-1,2,4-triazole. Acta Crystallogr C Cryst Struct Commun 48:1682–1683 https://doi.org/10.1107/S010827019100375X
Meng S, Zhao Y, Xue J, Zheng X (2018) Environment-dependent conformation investigation of 3-amino-1,2,4-triazole (3-AT): Raman Spectroscopy and density functional theory. Spectrochim Acta A Mol Biomol Spectrosc 190:478–485 https://doi.org/10.1016/j.saa.2017.09.053
Kubota S, Uda M (1975) 1,2,4-Triazoles. IV. Tautomerism of 3,5-disubstituted 1,2,4-triazoles. Chem Pharm Bull 23:955–966 https://doi.org/10.1248/cpb.23.955
Dolzhenko AV, Pastorin G, Dolzhenko AV, Keung Chui W (2009) An aqueous medium synthesis and tautomerism study of 3(5)-amino-1,2,4-triazoles. Tetrahedron Lett 50:2124–2128 https://doi.org/10.1016/j.tetlet.2009.02.172
Oziminski WP, Dobrowolski JCz, Mazurek AP, (2004) DFT studies on tautomerism of C5-substituted 1,2,4-triazoles. J Mol Struct Theochem 680:107–115 https://doi.org/10.1016/j.theochem.2004.05.005
Sergeieva T, Bilichenko M, Holodnyak S, Monaykina YV, Okovytyy SI, Kovalenko SI, Voronkov E, Leszczynski J (2016) Origin of substituent effect on tautomeric behavior of 1,2,4-triazole derivatives: Combined spectroscopic and theoretical study. J Phys Chem A 120:10116–10122 https://doi.org/10.1021/acs.jpca.6b08317
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery Jr JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V,Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09, Revision A.01. Gaussian Inc., Wallingford
Voronkov E, Rossikhin V, Okovytyy S, Shatckih A, Bolshakov V, Leszczynski J (2012) Novel physically adapted STO##-3G basis sets. Efficiency for prediction of second-order electric and magnetic properties of aromatic hydrocarbons. Int J Quantum Chem 112:2444−2449 https://doi.org/10.1002/qua.23256
Allouche A-R (2011) Gabedit − a graphical user interface for computational chemistry softwares. J Comput Chem 32:174–182 https://doi.org/10.1002/jcc.21600
Bilyi AK, Kovalenko SI, Pryhodko OB, Emets TI (2013) Investigation of growth-stimulating activity of 2-heteryl[1,2,4]triazolo[1,5-c]quinazolines and products of their nucleophilic degradation. Zaporizhzhia medical journal 2:83–86 https://doi.org/10.14739/2310-1210.2013.2.15606
Acknowledgements
We thank the Ministry of Education and Science of Ukraine for financial support (grant number is 0122U001220).
Funding
This study was supported by the Ministry of Education and Science of Ukraine (grant 0122U001220).
Author information
Authors and Affiliations
Contributions
O.P. calculated and interpreted the data, S.O. interpreted the data and was a contributor in writing and editing the manuscript, L.S. interpreted the data and was a contributor in writing and editing the manuscript, E.V. developed STO##-3Gel basis set, K.S. provided UV/vis spectroscopy experimental data, and S.K. provided UV/vis spectroscopy experimental data and was a contributor in writing and editing the manuscript. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Pylypenko, O.O., Okovytyy, S.I., Sviatenko, L.K. et al. Tautomeric behavior of 1,2,4-triazole derivatives: combined spectroscopic and theoretical study. Struct Chem 34, 181–192 (2023). https://doi.org/10.1007/s11224-022-02057-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-022-02057-0