
Abstract
Quantum chemistry calculations were performed to compare the reactivity indexes obtained within the conceptual density functional theory of phenolic and allyl-phenolic molecules and investigated in order to elucidate their antioxidant activity. Selected molecules share allyl and OH phenolic moieties, which can donate hydrogen atoms to highly reactive oxidant species. As a result, they inhibit or decrease the oxidative cycle. The calculation of bond dissociation energy relates this capability, together with reactivity indexes of the radicals produced, in order to measure and compare their stability. These indexes indicate a clear difference between these sets of structures with higher stability manifested by the allyl-phenolic-produced radicals.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Antioxidants are frequently known as playing a role in the reduction of symptoms of aging, cardiovascular illness, and neurodegenerative diseases, such as Alzheimer’s and Parkinson’s. Nowadays, these compounds are considered to be almost a panacea, promoting certain misconceptions in popular culture [1]. The first action of antioxidants is to reduce or neutralize reactive oxygen species (ROS) and thus decrease their oxidative process. Typical examples of ROS are OH, OOH, and NOx radicals and O2 singlet. These chemicals produce the so-called oxidative stress: resulting from an imbalance between the production of this oxidative species and its quenching by living beings. However, several biological processes implicate ROS; for example, the reduction of phagocytosis and ribonucleotides [2,3,4]. Phenolic- and allyl-derived compounds represent the main types of antioxidant molecules in experimental and computational studies. Both are arising from several natural vegetable sources [5]. Apart from the rigidity of their aromatic rings, phenolic compounds can also act as antioxidants because of the presence of hydroxyl and allyl groups [6, 7]. In addition to manifesting various biological activities, phenolic compounds often play crucial roles in food processing [8] which can be found in [9, 10] the form of complexes with other food components, such as protein and lipids, via hydrogen-bonding interactions. These components lead to changes in the physicochemical properties of the latter; for example, solubility, thermal stability, and digestibility. Some recent computational studies have been carried out on hydroxycinnamic acids [4], quercetin, and edaravone (both with DFT benchmark) [11], as well as catechin [12], flavones and flavonids [13], gallic acid [14], myricetin [13], pyranine [15], oxygenated terpenoids [16], and quinazoline derivatives [17]. Likewise, there are excellent reviews written about phenolic antioxidants [18, 19].
Plants represent an essential source of antioxidants, which are also commonly imbibed in tea. Asia is the leading producer of tea, and, in China and Japan, it is extensively utilized in their traditional medicine [20]. In Japan, these are called Kampo drugs, and their use recently has a boost because they have been mixed in modern medicine [21]. Besides, the antiviral activities of phenolic compounds from natural sources have been studied. For example, several phenolic compounds exhibit anti-HIV [22], can accomplish anti-tobacco mosaic virus activities, or even inhibit chikungunya and dengue virus replication [23]. Interestingly in [24], activities of phenolic compounds against the encephalomyocarditis virus were evaluated, indicating the number of phenolic hydroxyl groups significantly affects antiviral activity. Substituents also affect the antiviral activity of the compounds. Moreover, the relative position of functional groups also plays a crucial role in viral inhibition activity. This paper focuses on molecules on phenolic and allyl functional groups, for which antioxidant activity has been reported [25] (see Chart 1 for the studied molecules). A few questions were proposed concerning these compounds. For example are the following: Which of these two functional groups increase antioxidant activity? Which is the better antioxidant? Does the presence of both enhance antioxidant capacity? Which moiety produces the most stable radical following an H atom donation? With these questions in mind, we calculated two proposed antioxidant mechanisms—hydrogen atom transfer and single electron transfer. The first one, which refers to hydrogen atom transfer (HAT) consists in that the antioxidant molecule donates an H atom to the ROS, neutralizing its reactivity. Bond dissociation enthalpy (BDE) measures this reactivity, which involves breaking the –H bond [26, 27]. For the present paper, we considered both results from the breakdown of the phenolic O-H bond and breakdown of the H-allyl bond. Generally, the computed BDE values can compare to the corresponding one from phenol, as a representative molecule.

Antioxidant molecules from Asian tea
The second antioxidant mechanism studied in this paper is the Single Electron Transfer (SET). In this mechanism, there is a complete electron donation to the free radical. The viability of the mechanism relates to ionization energy (IE): in the case of an effective antioxidant, this quantity has a low value. Results are also usually compared to that of phenol [26]:
Antioxidant molecules manifest two critical features—the viability of electron donation to the free radical and the stability, or low reactivity, of the new radical formed by the antioxidant mechanism. BDE helps to measure the first one, as in thermodynamic terms, it shows how easily radical hydrogen donation is carried out by the antioxidant molecule. The second is related to conceptual DFT parameters, such as IE, hardness, electronegativity, and electrophilicity, thus measuring efficiency in terms of electron donation–acceptation. Concerning the stability and low reactivity of the radicals produced, we have calculated DFT reactivity indexes in order to analyze the chemical behavior of these species and compare phenolic and allylic radicals with well-known oxidant and antioxidant radicals.
Computational details
We used Gaussian 09 software package for all computations [28]. Initial geometries were built using Avogadro molecular editor [29], thus generating the corresponding input file. All molecular geometries were optimized at the M05-2X [30, 31], employing the 6-311++G(d,p) basis set for all chemical species studied (non-radical, radical, anion, and cation). Frequency calculations were computed to characterize each of the neutral and radical species as minima over the potential energy surface and in order to evaluate the zero-point energy corrections included in BDE values. Calculated ionization energy (IE) and electron affinity (EA) were both vertical. In this way, IE = Ecation − Eneutral and EA = Eneutral − Eanion. Calculation of BDE for OH and H-allyl bond dissociations was as follows: BDE = Hr + HH + Hneutral. Here, Hr is the enthalpy of the radicals generated, HH is the enthalpy of a hydrogen atom, and Hneutral is the enthalpy of a neutral molecule. We also applied B3LYP [32, 33] and LC-wPBE functional [34,35,36] (see supporting information) and CBS-QB3 methodology in order to compare and improve BDE energies [37]. CBS-n methods (CBS for complete basis set) are composite methods consisting of a sequence of geometric optimizations, calculating frequencies with a large basis set. They are usually followed by a series of single-point calculations, using higher level methods. Then, changing from a medium to smaller basis set, they apply an asymptotic extrapolation to reduce the error produced by the shortening of the basis sets employed [38]. CBS-QB3 is the medium-level method for this family that employs B3LYP/6-311G(2d,d,p) for geometry and frequency calculation and then a series of MP2, MP4, and CCSD(T) single points, as implemented in Gaussian 09. Previous works by us and other authors [11, 39] show a negligible influence on BDE and conceptual DFT indexes on the use of solvent effects, such as PCM (polarized continuum model). For this reason, we did not calculate them.
The calculation for electrophilicity requires electronegativity and hardness. For an N electron system with a potential external v(r) and total energy E, the partial derivative of the energy, related to the number of electrons at a constant potential, defines electronegativity. In a finite difference approximation, this is equivalent as half of the sum of IE and EA [40]:
Chemical hardness calculation was according to the definition proposed by Parr and Pearson: differentiating chemical potential to the number of electrons, at constant energy potential. The latter can approximate to half of the difference between IE and EA [41]. In order to symmetrize with electronegativity, the product multiplies by one half, as noted by Pearson [42]:
The calculation for the electrophilicity index is as:
IE and EA can also be used to calculate the electrodonor (ω−) and electroacceptor (ω+) indexes as proposed by Gázquez and co-workers [43]. Electrodonating power measures the propensity of donating charge, as defined by the following:
whereas the definition for electroaccepting power or the propensity to accept electrons (ω+) is as follows:
Low values for ω− are understood to indicate a greater capacity to donate charge, whereas high values for ω+ imply the greater capacity to accept a charge.
Plotting electrodonating and electroaccepting powers results in a donor–acceptor map, DAM, as proposed by Martínez [44]. This map, normalized with F as a model for suitable electron acceptor, and Na as a good electron donor, gives a useful graphic indicator. This comparison was therefore performed using computational values for F and Na atoms, at the same level of theory for the molecules studied. Thus, for any compound L, electron acceptance index is defined as follows:
If Ra = 1, L is a compound with an electron acceptor efficiency similar to F. If Ra > 1, L represents a more effective electron acceptor than F. Finally, if Ra < 1, L represents a less effective electron acceptor than F. Similarly, electron donation index is defined as follows:
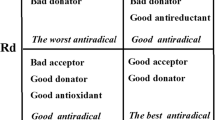
If Rd = 1, then L represents an electron donor with similar efficiency to Na. If Rd > 1, L is a less effective electron donor than Na. Likewise, if Rd < 1, L is a more effective electron donor than Na. Plotting Ra and Rd is the right way of visualizing the antioxidant scheme using MAP (Fig. 1). The graph has four central regions: the worst antiradical activity locates within the zone with poor donor and acceptor capacity. Two regions with good antiradical behavior correspond to bad acceptor but good donator and suitable acceptor but bad donator. Finally, the best antiradical zone stands for both suitable acceptor and donator.

Donor acceptor map (DAM) diagram. Four regions are distinguished and described in detail by Martinez. Dashed lines separating regions are only indicative, to clarify the image
Results
To accompany the study, we choose a couple of representative structures to compare the antioxidant capacity of the rest of the molecules. Lower limit corresponded to phenol, while the upper one to 2,2-diphenyl-picrylhydrazyl (DPPH). This molecule produces a very stable radical that is very well-known in antioxidant assays. The stabilization of DPPH radical is due to the distribution of the free electron over the whole molecule. The resulting species are hard to dimerize, which is contrary to most of the radicals [5]. Other molecules suggested for comparison are 2-propenyl-benzene and 1-propenyl-benzene. These molecules contrast with the collection proposed as they are non-phenolic compounds. The main structural difference in the case of these allyl and phenolic moieties is the non-planarity of the allyl moiety and phenolic ring. The latter can thus refer to as poor stabilization on the part of the unpaired electron between the ring and the allyl functional group. However, as shown in the subsequent texts, the calculated antioxidant parameters for allylic molecules are very similar and even better than the phenolic ones.
BDE of studied molecules
One of the most commonly utilized methods for defining antioxidant activity in computational chemistry is the calculation of BDE. This parameter can display the ability of electron and hydrogen donation in terms of the facility with which the X-H bond can break. A lower value for this energy indicates an increase in the possibility of hydrogen donation, resulting in the stabilization of other radicals in the oxidant cycle. The functionals and CBS-QB3 methods utilized yield a very similar tendency for BDE. The presentation of numerical results for calculations is in Table 1. Notably, recommending experimental BDE for phenol is 88.3 kcal mol−1 [45], and CBS-QB3 and M05-2X are the methods with values nearest to this. M05-2X also proved to be an excellent functional alternative to expensive methods, as it is similar to CBS-QB3 and experimental values are available. Moreover, it has been tested and benchmarked, particularly for antioxidant activity, and for the reaction kinetics of radicals [46]. In general, both B3LYP and LC-wPBE have similar values and are lower by approximately 3 kcal mol−1, when compared to M05-2X or CBS-QB3. Values for M05-2X and CBS-QB3 have a disparity to hydrogens from allyl moieties, by approximately 3 kcal mol−1. The calculation of BDE for simple allyl phenols presented for comparison purposes concurs well with experimental values, with 88.4 kcal mol−1 for prop-2-enylbenzene and 78.9 kcal mol−1 for (E)-1 phenylpropene [47]. In contrast, the results presented here for 1-phenylpropene are higher than those for prop-2-enylbenzene. However, experimental values represent the mean or average energies of the broken hydrogen bonds attached to the functional groups and can scarcely compare to energies calculated accurately here. BDE results are higher for simple phenolic compounds, such as phenol, p-ethyl-phenol, p-methyl phenol, or guaiacol. Considering the phenolic diol systems, the hydrogen bond formation between adjacent oxygens has lower BDE for catechol structures induced by a hydrogen bond formation [48]. Similarly, the presence of allyl or extra phenyl functional groups attached to a phenolic ring produces a lower BDE than that of phenol, for at least one type of hydrogen. Interesting cases are guaiacol and eugenol, for which BDE for allyl hydrogen decreases. The experimental data we consulted from TBARS corroborates our finding that the presence of the allyl group increases antioxidant capacity [25]. In these reports, the tendency for antioxidant activity is 4-allyl-2,6-dimethoxyphenol > 4_4biphenol > eugenol > 2-allyl-6-methyl_phenol > honokiol > magnolol > p-ethylphenol > guaiacol, as made apparent in Table 1 by the percentage of lipid oxidation inhibition. A possible explanation regarding the lower values for allyl hydrogen BDE compared to those of hydroxyl groups refers to the difference in electronegativity between carbon and oxygen. The latter appears to be more plausible than stabilization of the radical because of geometrical (planar) reasons. The geometry of the allyl in non-radical and radical structures has poor coplanarity with the aromatic ring (XYZ coordinates of the molecules studied are available in the Supplementary Information). In this way, electronic delocalization of the unpaired electron in the yielded radical seems unsuitable.
IE of antioxidants
Ionization energy is a popular tool for measuring antioxidant capacity utilized in computational chemistry (Table 2) [26]. This index may indicate the possibility of an electron donation from the antioxidant to highly oxidative species, thus breaking the oxidant cycle. Usually, it compares to that of phenol as a representative phenolic molecule. Lower IE means greater capacity for electron donation. We also calculated the IE of DPPH and used this as an example of a good antioxidant molecule. DPPH can produce a very stable radical, and its properties may be useful for contrasting the antioxidant properties of the studied set of molecules [49]. The tendency is similar to BDE: that is, simple phenolic systems, such as phenol and allylphenols, have the highest IE. The representative antioxidant, DPPH, has an IE of 8.1 eV; therefore, molecules with a similar or lower IE title as good electron donors. Honokiol is the molecule with less IE, followed by 4-prop-1-enyl benzene-1,2-diol 6; 4–4′-biphenol, 4-allyl-2,6-dimethoxy phenol; and 2allyl-6-methoxy phenol. The rest of the studied molecules are not as efficient electron donors as DPPH; however, they are more efficient compared to phenol or simple allylphenols.
EA of antioxidants
Results for EA are in Table 2. The representative antioxidant, DPPH, has the highest EA with 2.0 eV, whereas the lowest values are for phenol, p-ethylphenol, and p-methyl phenol with − 1.4 eV, followed by allyl phenols (− 0.8 eV). Magnolol has a calculated EA of − 0.4 eV whereas, for the rest of molecules, EA values are close to − 0.7 eV. EA can clarify the electron-accepting behavior of a molecule. In this case, pure phenolic compounds are bad at accepting electrons whereas, in contrast, DPPH manifests good electron-accepting behavior, indicated by its positive EA. Due to the negative values of EA, the molecules studied are not suitable electron acceptors. The latter is made evident in the donor–acceptor map displayed in the subsequent texts.
Conceptual DFT indexes of antioxidants
Hardness, electronegativity, and electrophilicity are also in Table 2. Hardness measures the resistance on the part of the chemical potential to change the number of electrons [42]. The latter means that phenol with the greatest hardness is less likely to change electronic distributions, and it is also the structure that manifests least antioxidant activity. Likewise, DPPH is the least hard molecule, thus increasing the possibility that will change its electronic distribution, confirming that this molecule is a more efficient antioxidant. There is a tendency for the simple phenolic structures like allylbenzenes, guaiacol, p-ethylphenol, and p-methyl phenol to manifest hardness similar to that of phenol (higher than 4.7 eV), whereas the rest of the studied structures have low values. Therefore, a lower hardness than phenol characterizes antioxidant activity for these molecules. There is a noticeable relationship between the number of hydroxyl groups and hardness values, as apparent in Table 2. Excepting DPPH and phenol compounds (which are the extreme values), the lowest values for hardness belong to honokiol, 4-prop-1-enyl benzene-1,2-diol, and 4–4′-biphenol, which have two hydroxyl groups. In contrast, the highest values of hardness belong to 2-propenyl benzene and ethylphenol, which have zero and a single OH group. This index may relate to the relationship between the number of hydroxyl groups and antiviral activity reported in Refs. [22, 24]. Electronegativity values do not tend in the way hardness does. However, interestingly, DPPH has the highest value, and there is no clear difference concerning the rest of the structures studied. There is also no relationship between the OH number and this index, as evidenced in Table 2: honokiol has two OH groups and has the lowest value for electronegativity. Likewise, ethylphenol, p-methylphenol, and 2allyl-6-methoxy phenol have a single OH and low values for hardness.
In contrast, compounds with the highest values of electronegativity, including DPPH, are 2-propenyl benzene, magnolol, and guaiacol, which have zero, two, and one OH groups, respectively. For the set of molecules studied, electronegativity may not be the best tool for analyzing these structures. A similar case shows electrophilicity, which is the highest value obtained for DPPH, with no apparent difference to the rest of the structures studied. Relatively high electronegativity and electrophilicity, in the case of phenol, may be indicative of a good antioxidant. However, there is no significant difference between these values and the molecules studied.
Donor–acceptor maps for antioxidants
The values plotted for the set of compounds studied (Fig. 2) place them in the zone of good antiradical behavior with the suitable donor but bad electron-accepting capacity. The value for Rd slightly above 1 shows a capacity of electron donation superior to the Na atom [44]. The set of molecules studied has very similar DAM values, even approaching that of phenol. In contrast, DPPH occurs as a molecule with one of the best antiradical behavior; that is, good electron donor and acceptor capacities. Low values of hardness also corroborate this dual behavior on the part of DPPH.

Donor–acceptor map for molecules studied during the gas phase. Points on the left, most of them hard to distinguish, are the antioxidants studied here and classify as good antiradical. The isolated point to the right represents DPPH shown to be one of the best radicals according to the scheme proposed by Martínez
Radicals
The low reactivity of the produced radical is also a characteristic of a good antioxidant. Calculation of conceptual DFT parameters for the formed radicals helped to analyze this. Radicals derived from phenol, propenyl benzene, and allylbenzene served as examples of low antioxidant activity, whereas the radical obtained from DPPH was an example of a good antioxidant. Other attempts to clarify the nature of radicals by the conceptual DFT were reported, for example, the comparison of these parameters with that of molecules previously assumed to have low reactivity [50] or analyzing a broad set of structures in order to build a scale of reactivity and a particular property, such as hardness or electrophilicity [51, 52]. The main goal of this paper is to analyze radicals of molecules with the phenolic and allylbenzene moieties and compare their stability via conceptual DFT parameters. The calculated numerical data shows a significant difference between phenolic and allylbenzene radicals with low reactivity in the case of these last ones. Concerning ionization energy in Fig. 3, the analyzed radicals show similar values to the parent non-radical structures. This energy indicates the possibility of obtaining an extra electron from radical to oxidative species. The highest IE value is for phenol radical with 9.0 eV. The stable radical from DPPH has a slightly higher IE than benzene allyl radicals, which have the lowest values of any molecules studied. Highly reactive radicals, such as OOH and OH, were reported previously to have higher IE (12.8 and 16.2 eV, respectively) [39]. Thus, for the set of molecules studied, a low IE is indicative of a radical with low reactivity. A similar trend occurs with EA (Fig. 3), lower values by approximately 1 eV for allyl radicals compared to phenolic ones (close to 2 eV). In this case, DPPH has shown a similar EA to phenol (2.2 and 2.7 eV, respectively).

Graph of ionization energy (full squares) and electron affinity (open squares) for the studied radicals. The labels on the x-axis correspond to those in Chart 1 and Table 1. DPPH radical label is 15. The tendency is as follows: lower values for allyl radicals and higher ones for phenolic radicals, for both properties
Concerning hardness (Fig. 4), there is no significant difference between allylic and phenolic radicals. However, it is noticeable that DPPH has the lowest values, whereas phenol has the highest ones (3.4 and 4.4 eV, respectively) for this index. This information referring to the stability and low reactivity of DPPH contrasts with oxidative OOH and OH radicals, with hardness values of 6.3 and 7.2 eV. Likewise, electronegativity (Fig. 4) shows a similar tendency to IE, with a difference of approximately 1 eV between phenolic and allylic radicals with the highest value for phenol (5.6 eV). Low electronegativity characterizes to low reactivity radicals, in comparison to oxidant radicals OOH and OH, which have an electronegativity of 6.6 and 8.9 eV, less than that of phenol.

Graph showing electronegativity (squares) and hardness (circles) for radicals studied. The labels on the x-axis correspond to those of Chart 1 and Table 1. DPPH radical label is 15. Concerning electronegativity, the tendency is as follows: lower values for allyl radicals and higher ones for phenolic radicals. There is no clear tendency for hardness
The electrophilicity (Fig. 5) for the radicals studied also shows a difference between allylic and phenolic radicals of approximately 1 eV. These values were even more in some cases, with the allyl radicals showing the lowest values. DPPH and phenol have relatively high electrophilicity values. The latter indicates that allylic radicals have insufficient capacity for accepting more electrons, especially compared to phenyl radicals or even DPPH radicals. Highly reactive radical OH has same electrophilicity as DPPH (5.5 eV), whereas OOH has a lower value (3.6 eV). As there is no apparent difference between the radicals studied and prototype molecules, electrophilicity is not a very good indicator for clarifying tendencies among the radicals studied.

Frontier molecular orbitals
The study of the antioxidant properties of the molecules was also made analyzing the highest occupied molecular orbital HOMO. The lower the energy in this orbital, the higher the possibility to donate an electron. Also, qualitatively, the electron density of HOMO can offer a view of the electron-donating site region. As a complement, the electron density of LUMO can show the electron-accepting area of high oxidative radicals in the molecule. The HOMO values for the molecules studied here (Fig. 6) cannot be entirely associated with their antioxidant character, as this should depend on the specific reaction. For example, magnolol 11 manifests a high percentage of lipid oxidation inhibition (see Table 1). However, it presents the lowest HOMO values (− 7.6 eV), which are very similar to those of other compounds, such as p-ethylphenol 7 and p-methylphenol 12 (− 7.5 eV). They are known to be weak oxidation inhibitors. Even eugenol 8 (− 7.5 eV), which can be cataloged as a weak antioxidant, shows a good percentage of lipid oxidation inhibition. These values imply that HOMO values may still manifest good antioxidant behavior on the part of molecules, despite the relatively high energy of this orbital. It also shows that they represent good antioxidants in terms of their percentage of lipid oxidation inhibition. From a qualitative perspective, HOMO is mainly located over aromatic rings and hydroxyl moiety but concentrates poorly over the allyl group, with 4-prop-1-enylbenzene-1,2-diol 6 representing the only exception. The poor coplanarity of the allyl and benzene fragments explains this exception. As a result, there is a lower extension of electronic delocalization of the double bond near the benzene cycle. Concerning the electron density of the HOMO of the hydroxyl group, this may show the region of the available electron or H atom from this and the consequent stabilization of the yielded radical by the aromatic ring. Contrastingly, LUMO locates mainly over the allyl group, aromatic ring, and the rest of the organic functional groups. This location characterizes zones of the molecules in a way similar to electron acceptors. The difference between the hydroxyl group as an electron donor and the allyl as an electron acceptor is apparent.

Graphs showing frontier molecular orbitals for molecules studied. Structures are numbering as in Chart 1. Values are in eV
Conclusion
The following facts should be emphasized for the studied molecules:
HAT mechanism for allyl-type hydrogen displays an antioxidant capacity, which is as good as that of hydroxyl-phenolic, showing similar BDE values or even lower ones in the case of some allyl compounds.
IE tends to show lower values for molecules with more functional groups attached to the aromatic ring. It is well-established that electron donor groups reduce the IE of catechols [53]. In this study, both allyl and hydroxy groups are the functional groups of electron donors. The latter allows us to associate that the reduction of IE is mainly influenced by this fact, as evident in the results for ionization energy. Moreover, allyl-benzene compounds and phenol tend to have high IE values.
Concerning DFT indexes for molecules, interestingly DPPH is characterized by low hardness, high electronegativity, and high electrophilicity. Similarly, IE and EA show DPPH to be a good electron acceptor–donor. In contrast, phenol exhibits high hardness, low electronegativity, and low electrophilicity. Bearing this in mind, all of the antioxidants studied manifest good antioxidant activity. However, as DFT indexes present very similar values for the molecule set, it is difficult to discern or detect a tendency. Notwithstanding, all antioxidants studied manifest good antioxidant activity.
Concerning the radicals yielded following hydrogen donation, DFT indexes for allyl radical show lower IE, electronegativity, and electrophilicity than hydroxy-benzene radicals. Comparing these compounds to highly oxidative radicals, such as OOH and OH that have relatively high IE and electronegativity, the radicals studied show poor reactivity and therefore good stability, with allyl radicals being the most stable. Likewise, the hardness of radicals studied is low compared to OOH and OH, corroborating the idea of the stability of the radicals analyzed.
References
Bast A, Haenen GRMM (2013) Ten misconceptions about antioxidants. Trends Pharmacol Sci 34:430–436. https://doi.org/10.1016/j.tips.2013.05.010
Azevedo J, Oliveira J, Cruz L et al (2014) Antioxidant features of red wine pyranoanthocyanins: experimental and theoretical approaches. J Agric Food Chem 62:7002–7009. https://doi.org/10.1021/jf404735j
Vargas-Sánchez RD, Mendoza-Wilson AM, Torrescano-Urrutia GR, Sánchez-Escalante A (2015) Antiradical potential of phenolic compounds fingerprints of propolis extracts: DFT approach. Comput Theor Chem 1066:7–13. https://doi.org/10.1016/j.comptc.2015.05.003
Mazzone G, Russo N, Toscano M (2016) Antioxidant properties comparative study of natural hydroxycinnamic acids and structurally modified derivatives: computational insights. Antioxidants vs Oxidative Stress Insights from Comput 1077:39–47. https://doi.org/10.1016/j.comptc.2015.10.011
Chen Y, Xiao H, Zheng J, Liang G (2015) Structure-thermodynamics-antioxidant activity relationships of selected natural phenolic acids and derivatives: an experimental and theoretical evaluation. PLoS One 10:e0121276. https://doi.org/10.1371/journal.pone.0121276
Yang G, Kristufek SL, Link LA et al (2016) Thiol–ene elastomers derived from biobased phenolic acids with varying functionality. Macromolecules 49:7737–7748. https://doi.org/10.1021/acs.macromol.6b01018
Yang G, Kristufek SL, Link LA et al (2015) Synthesis and physical properties of thiol–ene networks utilizing plant-derived phenolic acids. Macromolecules 48:8418–8427. https://doi.org/10.1021/acs.macromol.5b01796
Ozdal T, Capanoglu E, Altay F (2013) Review. Food Res Int 51:954–970. https://doi.org/10.1016/j.foodres.2013.02.009
Jung D-M, de Ropp JS, Ebeler SE (2000) Study of interactions between food phenolics and aromatic flavors using one- and two-dimensional 1H NMR spectroscopy. J Agric Food Chem 48:407–412. https://doi.org/10.1021/jf9906883
Viljanen K, Kivikari R, Heinonen M (2004) Protein−lipid interactions during liposome oxidation with added anthocyanin and other phenolic compounds. J Agric Food Chem 52:1104–1111. https://doi.org/10.1021/jf034785e
La Rocca MV, Rutkowski M, Ringeissen S et al (2016) Benchmarking the DFT methodology for assessing antioxidant-related properties: quercetin and edaravone as case studies. J Mol Model 22:250. https://doi.org/10.1007/s00894-016-3118-6
Wang J, Tang H, Hou B et al (2017) Synthesis, antioxidant activity, and density functional theory study of catechin derivatives. RSC Adv 7:54136–54141. https://doi.org/10.1039/C7RA11496F
Sroka Z, Zbikowska B, Hladyszowski J (2015) The antiradical activity of some selected flavones and flavonols. Experimental and quantum mechanical study. J Mol Model 21. https://doi.org/10.1007/s00894-015-2848-1
Badhani B, Kakkar R (2017) DFT study of structural and electronic properties of gallic acid and its anions in gas phase and in aqueous solution. Struct Chem 28:1789–1802. https://doi.org/10.1007/s11224-017-0958-3
Aspée A, Aliaga C, Maretti L et al (2017) Reaction kinetics of phenolic antioxidants toward photoinduced pyranine free radicals in biological models. J Phys Chem B 121:6331–6340. https://doi.org/10.1021/acs.jpcb.7b02779
Ngo TC, Dao DQ, Nguyen MT, Nam PC (2017) A DFT analysis on the radical scavenging activity of oxygenated terpenoids present in the extract of the buds of Cleistocalyx operculatus. RSC Adv 7:39686–39698. https://doi.org/10.1039/C7RA04798C
Lakshmanan S, Govindaraj D, Ramalakshmi N, Antony SA (2017) Synthesis, molecular docking, DFT calculations and cytotoxicity activity of benzo[g]quinazoline derivatives in choline chloride-urea. J Mol Struct 1150:88–95. https://doi.org/10.1016/j.molstruc.2017.08.082
Alov P, Tsakovska I, Pajeva I (2015) Computational studies of free radical-scavenging properties of phenolic compounds. Curr Top Med Chem 15:85–104. https://doi.org/10.2174/1568026615666141209143702
Shahidi F, Ambigaipalan P (2015) Phenolics and polyphenolics in foods, beverages and spices: antioxidant activity and health effects—a review. J Funct Foods 18:820–897. https://doi.org/10.1016/j.jff.2015.06.018
Suzuki T, Miyoshi N, Hayakawa S et al (2016) Health benefits of tea consumption. In: Wilson T, Temple NJ (eds) Beverage impacts on health and nutritionSecond edn. Springer International Publishing, Cham, pp 49–67
Tanaka J (2012) Japanese tea breeding history and the future perspective. In: Chen L, Apostolides Z, Chen Z-M (eds) Global tea breeding: achievements, challenges and perspectives. Springer Berlin Heidelberg, Berlin, pp 227–239
Hu Q-F, Zhou B, Huang J-M et al (2013) Antiviral phenolic compounds from Arundina gramnifolia. J Nat Prod 76:292–296. https://doi.org/10.1021/np300727f
Bourjot M, Leyssen P, Eydoux C et al (2012) Flacourtosides A–F, phenolic glycosides isolated from Flacourtia ramontchi. J Nat Prod 75:752–758. https://doi.org/10.1021/np300059n
Li R, Narita R, Nishimura H et al (2018) Antiviral activity of phenolic derivatives in pyroligneous acid from hardwood, softwood, and bamboo. ACS Sustain Chem Eng 6:119–126. https://doi.org/10.1021/acssuschemeng.7b01265
Ogata M, Hoshi M, Shimotohno K et al (1997) Antioxidant activity of magnolol, honokiol, and related phenolic compounds. J Am Oil Chem Soc 74:557–562. https://doi.org/10.1007/s11746-997-0180-3
Wright JS, Johnson ER, DiLabio GA (2001) Predicting the activity of phenolic antioxidants: theoretical method, analysis of substituent effects, and application to major families of antioxidants. J Am Chem Soc 123:1173–1183. https://doi.org/10.1021/ja002455u
Zhang H-Y, Sun Y-M, Wang X-L (2003) Substituent effects on O–H bond dissociation enthalpies and ionization potentials of catechols: a DFT study and its implications in the rational design of phenolic antioxidants and elucidation of structure–activity relationships for flavonoid antioxidants. Chem Eur J 9:502–508. https://doi.org/10.1002/chem.200390052
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery Jr JA, Peralta JE, Ogliaro F, Bearpark MJ, Heyd J, Brothers EN, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam NJ, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09. Gaussian, Inc., Wallingford
Hanwell MD, Curtis DE, Lonie DC, et al (2012) Avogadro: an advanced semantic chemical editor, visualization, and analysis platform
Zhao Y, Schultz NE, Truhlar DG (2006) Design of density functionals by combining the method of constraint satisfaction with parametrization for thermochemistry, thermochemical kinetics, and noncovalent interactions. J Chem Theory Comput 2:364–382. https://doi.org/10.1021/ct0502763
Zhao Y, Truhlar DG (2008) How well can new-generation density functionals describe the energetics of bond-dissociation reactions producing radicals? J Phys Chem A 112:1095–1099. https://doi.org/10.1021/jp7109127
Becke AD (1988) Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A 38:3098–3100
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789
Vydrov OA, Scuseria GE (2006) Assessment of a long-range corrected hybrid functional. J Chem Phys 125:234109. https://doi.org/10.1063/1.2409292
Vydrov OA, Heyd J, Krukau AV, Scuseria GE (2006) Importance of short-range versus long-range Hartree-Fock exchange for the performance of hybrid density functionals. J Chem Phys 125:74106. https://doi.org/10.1063/1.2244560
Vydrov OA, Scuseria GE, Perdew JP (2007) Tests of functionals for systems with fractional electron number. J Chem Phys 126:154109. https://doi.org/10.1063/1.2723119
Somers KP, Simmie JM (2015) Benchmarking compound methods (CBS-QB3, CBS-APNO, G3, G4, W1BD) against the active thermochemical tables: formation enthalpies of radicals. J Phys Chem A 119:8922–8933. https://doi.org/10.1021/acs.jpca.5b05448
Montgomery JA, Frisch MJ, Ochterski JW, Petersson GA (2000) A complete basis set model chemistry. VII. Use of the minimum population localization method. J Chem Phys 112:6532–6542. https://doi.org/10.1063/1.481224
Hernandez DA, Tenorio FJ (2017) Reactivity indexes of antioxidant molecules from Rosmarinus officinalis. Struct Chem. https://doi.org/10.1007/s11224-017-1066-0
Parr RG, Donnelly RA, Levy M, Palke WE (1978) Electronegativity: the density functional viewpoint. J Chem Phys 68:3801–3807. https://doi.org/10.1063/1.436185
Parr RG, Pearson RG (1983) Absolute hardness: companion parameter to absolute electronegativity. J Am Chem Soc 105:7512–7516. https://doi.org/10.1021/ja00364a005
Pearson RG (2005) Chemical hardness and density functional theory. J Chem Sci 117:369–377. https://doi.org/10.1007/BF02708340
Gázquez JL, Cedillo A, Vela A (2007) Electrodonating and electroaccepting powers. J Phys Chem A 111:1966–1970. https://doi.org/10.1021/jp065459f
Martínez A (2009) Donator acceptor map of psittacofulvins and anthocyanins: are they good antioxidant substances? J Phys Chem B 113:4915–4921. https://doi.org/10.1021/jp8102436
Dorofeeva OV, Ryzhova ON (2016) Enthalpy of formation and O–H bond dissociation enthalpy of phenol: inconsistency between theory and experiment. J Phys Chem A 120:2471–2479. https://doi.org/10.1021/acs.jpca.6b02233
Galano A, Alvarez-Idaboy JR (2014) Kinetics of radical-molecule reactions in aqueous solution: a benchmark study of the performance of density functional methods. J Comput Chem 35:2019–2026. https://doi.org/10.1002/jcc.23715
Luo YR (2002) Handbook of bond dissociation energies in organic compounds. CRC Press
Ortega-Moo C, Garza J, Vargas R (2016) The substituent effect on the antioxidant capacity of catechols and resorcinols. Theor Chem Accounts 135:177. https://doi.org/10.1007/s00214-016-1932-7
Yar M, Arshad M, Farooq A et al (2015) Synthesis and DPPH scavenging assay of reserpine analogues, computational studies and in silico docking studies in AChE and BChE responsible for Alzheimer’s disease. Braz J Pharm Sci 51:53–61. https://doi.org/10.1590/S1984-82502015000100006
Domingo LR, Perez P (2013) Global and local reactivity indices for electrophilic/nucleophilic free radicals. Org Biomol Chem 11:4350–4358. https://doi.org/10.1039/C3OB40337H
De Vleeschouwer F, Van Speybroeck V, Waroquier M et al (2007) Electrophilicity and nucleophilicity index for radicals. Org Lett 9:2721–2724. https://doi.org/10.1021/ol071038k
De Vleeschouwer F, Geerlings P, De Proft F (2012) Radical electrophilicities in solvent. Theor Chem Accounts 131:1–13. https://doi.org/10.1007/s00214-012-1245-4
Zhang H-Y, Wang L-F (2002) Are allylic hydrogens in catechins more abstractable than catecholic hydrogens? J Am Oil Chem Soc 79:943–944. https://doi.org/10.1007/s11746-002-0583-6
Acknowledgments
Financial support by PRODEP/DSA/511/18-9169 UDG-PTC-1436, CONACyT-Mexico by grant 52827, and ProSNI program of Universidad de Guadalajara is acknowledged. DAH thanks the “Programa de Repatriaciones y Retenciones 2017” of CONACyT-Mexico and Universidad de Guadalajara. The authors gratefully acknowledge the computing time granted by LANCAD and CONACYT on the supercomputer Yoltla/Miztli/Xiuhcoatl at LSVP UAM-Iztapalapa/DGTIC UNAM/CGSTIC CINVESTAV.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest regarding the publication of this article.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Hernandez, D.A., Rodriguez-Zavala, J.G. & Tenorio, F.J. DFT study of antioxidant molecules from traditional Japanese and Chinese teas: comparing allylic and phenolic antiradical activity. Struct Chem 31, 359–369 (2020). https://doi.org/10.1007/s11224-019-01411-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-019-01411-z