
Abstract
Three new coordination complexes of Zn(II) and Mn(II) have been synthesised using two different tridentate N,N,O donor hydrazone ligands, Hpbh and Hacpbh respectively. The complexes [Zn(pbh)2] (1) and [Zn(acpbh)2] (2) have been synthesized by the treatment of ZnSO4 · 7H2O with Hpbh and Hacpbh hydrazone ligands, respectively. The Mn(II) complex [Mn(acpbh)2] (3) was obtained on reacting Mn(NO3)2 · 4H2O with the ligand Hacpbh. The ligands Hpbh and Hacpbh were prepared by condensing pyridine-2-carboxaldehyde and 2-acetylpyridine with benzhydrazide respectively. Inspite of varying the carbonyl functionality attached to the pyridine moiety present in the hydrazone ligands in both the Schiff bases, we obtained three mononuclear complexes 1, 2, and 3 which were clearly characterized from single crystal X-ray diffraction studies. Spectroscopic investigations like IR and UV/Vis have been carried out for 1, 2, and 3. Fluorescence studies have been performed for 1 and 2. For 3 cyclic voltammetry, room temperature magnetic study and EPR measurements have been recorded.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Arylhydrazone complexes of transition metal ions are known to provide useful models for ellucidation of the mechanisms of enzyme inhibition by hydrazine derivatives [1] and for their possible pharmacological applications [2]. The activity of some hydrazone complexes is very significant against Gram-positive bacteria in vitro. These hydrazone chelate derivatives act as good potential oral drugs to treat the genetic disorders like thalassemia [3]. The structural characterization of these resultant hydrazone complexes revealed some interesting facts, such as their tendency and potency to act as planar pentadentate ligands in most of the complexes [4–7] along with tridentate character [7–11]. Moreover, these ligands exhibit keto-enol tautomerism (Fig. 1) and can coordinate in neutral [12], monoanionic [13], dianionic [4, 14, 15] or tetraanionic [16] forms, to the metal ions which have coordination numbers of six and seven [4, 17]; generating mononuclear or binuclear species. However, it depends on the reaction conditions, such as metal ion, its concentration, the pH of the medium, and the nature of the hydrazone ligand [13]. Zinc(II) ion has been widely found in several zinc containing metalloenzymes such as zinc-peptidases [18], human carbonic anhydrase [19], and alkalinephosphatase [20]. The coordination mode for octahedral or tetrahedral geometries of zinc(II) ion may affect the formation and mechanism of the self-assembly processes. The chemistry of manganese, in various oxidation states and in various combinations of nitrogen and oxygen donor environment, is presently witnessing intense activity [21–24]. This is primarily due to the role of manganese in various biological processes, e.g., in the enzymes such as pseudocatalase [25–27], mitochondrial superoxide dismutase [28], and most significantly in the water oxidation complex (WOC) in photosystem-II [29, 30].

Keto-enol tautomerism of hydrazone ligands
In this article we describe the synthesis, structural ellucidation, fluorescence study of two Zn(II) complexes 1 and 2. For the Mn(II) complex 3 in addition to synthesis, structural ellucidation, EPR spectra, magnetic study and electrochemical properties have also been described. The molecular structures of 1, 2, and 3 have also been established from single crystal X-ray diffraction study which show that in all the three complexes the transition metal ion is in N4O2 donor environment coordinated via pyridine–N, imine–N and deprotonated amide–O of the two different hydrazone ligands Hpbh and Hacpbh, respectively.
Experimental
Materials
All chemicals and solvents used for the syntheses were of analytical grade. Pyridine-2-carboxaldehyde, 2-acetylpyridine, benzhydrazide, zinc sulphate heptahydrate, manganese nitrate tetrahydrate were all purchased form Aldrich Chemical Co. and were used without further purification.
Preparation of the ligands and the complexes
Synthesis of the Schiff base ligands: Hpbh and Hacpbh
The Schiff base ligands (Hpbh and Hacpbh) were prepared according to the reported methods [31–33]. The two tridentate Schiff bases Hpbh and Hacpbh were prepared by the reflux condensation of benzhydrazide with pyridine-2-carboxaldehyde and 2-acetylpyridine in (1:1 mmol ratio) respectively. The former was refluxed for 2 h and the later for 15 h which then gave yellow-colored solutions. The Schiff bases were used without further purifications.
Synthesis of 1
A total of 20 cm3 of a methanolic solution of the Schiff base ligand (Hpbh) (0.45 g, 2 mmol), was added drop wise to a vigorously stirred warm 10 cm3 methanolic ZnSO4 · 7H2O (0.383 g, 1 mmol) solution. The resulting solution was vigorously stirred at 40 °C for 1 h. After filtration the filtrate was kept at 26 °C, when pale yellow single crystals suitable for X-ray diffraction appeared after one night standing.
Yield: 0.3255 gm, 85% with respect to the zinc salt.
Anal. Calc. (%) for C26 H20 N6 O2 Zn: C, 60.77; H, 3.92; N, 16.35. Found (%): C, 60.72; H, 3.89; N, 16.33.
Synthesis of 2
ZnSO4 · 7H2O (0.383 g, 1 mmol) was dissolved in 10 cm3 methanol and to it 20 cm3 methanolic solution of the Schiff base (Hacpbh) (0.47 g, 2 mmol), was added dropwise in stirring condition. After the addition was complete, the solution was vigorously stirred at 40 °C for 2 h. The solid obtained was filtered, washed several times with methanol followed by ether and then dried over fused CaCl2. Pale yellow rectangular shaped single crystals of 2 suitable for X-ray diffraction were obtained by diffusion of a dichloromethane solution of the complex into hexane.
Yield: 0.2834 gm, 74% with respect to the zinc salt.
Anal. Calc. (%) for C28 H24 N6 O2 Zn: C, 62.06; H, 4.46; N, 15.51. Found (%): C, 62.03; H, 4.44; N, 15.49.
Synthesis of 3
Mn(NO3)2 · 4H2O (0.251 g, 1 mmol) was dissolved in 10 cm3 methanol. 20 cm3 pale yellow methanolic solution of the Schiff base (Hacpbh) (0.47 g, 2 mmol), was added to the metal salt solution dropwise and the resulting solution was stirred. On addition of Et3N (0.202 g, 2 mmol) to the above mixture, an immediate reddish brown solid separated. The mixture was further stirred for 2 h. The solid obtained was filtered, washed several times with methanol followed by ether and then dried over fused CaCl2. Single crystals of 3 suitable for X-ray diffraction were obtained by diffusion of a dichloromethane solution of the complex into hexane.
Yield: 0.1907 gm, 76% with respect to the manganese salt.
Anal. Calc. (%) for C29 H28 Mn N6 O3: C, 61.81; H, 5.01; N, 14.91. Found (%): C, 61.79; H, 4.98; N, 14.87.
Physical measurements
The Fourier Transform Infrared spectra of the complexes were recorded on a Perkin–Elmer Spectrum RX I FT-IR in the range 4,000–300 cm−1 system with solid KBr disc. The electronic spectra were recorded at 300 K on a Perkin–Elmer λ-40 UV/Vis-spectrometer using HPLC grade acetonitrile as solvent with a 1-cm quartz cubates in the range 200–800 nm. C, H, N microanalyses were carried out with a Perkin–Elmer 2400 II elemental analyzer. Electrochemical measurements were performed on a CH 600A cyclic voltametry instrument using HPLC grade acetonitrile solution as solvent where tetrabutylammonium perchlorate was used as supporting electrolyte at a scan rate 0.05 V/s. Platinum and standard calomel electrode (SCE) were the working and the reference electrode in the process respectively. Room temperature magnetic susceptibility was measured in solid phase with a Sherwood-Scientific, Cambridge, UK magnetic susceptibility balance using conventional Gouy’s method taking mercury tetrathiocyanatocobaltate(II) as standard. Diamagnetic corrections were done using Pascal’s Table. X-band Electron Paramagnetic Resonance spectra were recorded with a Bruker ER200D spectrometer equipped with an NMR-Gaussmeter, an Anritsu microwave frequency counter, and an Oxford ESR9 cryostat. JOBIN YVON Fluoro Max 3. A spectroflurometer model was used for the steady state fluorescence measurements for both complexes (1 and 2) and the ligands (Hpbh and Hacpbh) at room temperature (298 K). The solutions of the ligands and the complexes were prepared in the ratio of 2:1 methanol:water mixed solvents and OD for each of the solutions at the wavelength for fluorescence excitation were recorded.
Single crystal X-ray diffraction study: data collection and structure refinement
The crystal structure analyses of 1, 2, and 3 were performed by three different authors in different laboratories. The equipment used for each sample is listed with the crystallographic data and the refinement results in Table 1. The data were reduced to F 2o and corrected for multiscan absorption effects with Bruker SHELXTL [34], SADABS [35] and CrysAlis PRO [36] softwares, respectively. Unit cell refinements were done using the CrysAlis PRO, Bruker SMART softwares [37]. For the structural refinement of complex 3, the SQUEEZE program was used to deal with the two disordered methanol molecules which were eliminated for a best refinement of the molecule [38]. The structures of all the complexes were determined by direct methods procedures in either a version of SHELXS [39] and refined by full-matrix least-squares methods in SHELXL [40]. All non hydrogen atoms were refined with anisotropic displacement parameters. All hydrogen atoms were located in calculated positions to correspond to standard bond lengths and angles.
Results and discussion
Infrared spectra
The IR spectra of 1, 2, and 3 were analyzed in comparison with that of their respective free ligands (Hpbh and Hacpbh) in the region 4,000–250 cm−1. The IR spectra of the hydrazone ligands contain a strong C–O absorption band at 1,651–1,659 cm−1 and a N–H absorption band at 3,187–3,250 cm−1. Both of these bands disappear on complexation, and a new C–O absorption band appears at 1,040–1,089 cm−1 in the complexes, indicating that the hydrazone ligands have undergone deprotonation on complexation. These data give evidence for the coordination of the ligands (Hpbh and Hacpbh) to the respective metal ions [Zn(II) and Mn(II)] via two nitrogen atoms and one oxygen atom. The infrared spectra of 1, 2 and 3 display IR absorption bands at 1,637, 1,583, and 1,587 cm−1 which can be assigned to the C=N stretching frequencies of the coordinated ligands (Hpbh and Hacpbh) [41], whereas for the free ligands (Hpbh and Hacpbh) the same bands are observed at 1,652 and 1,685 cm−1 respectively. The shift of these bands on complexation towards lower wave number indicate coordination of the azomethane nitrogen to the metal centre [42]. The ligand coordination to the metal centre is substantiated by two bands appearing at 412, 414, 416 cm−1 and at 380, 374, 360 cm−1 for 1, 2, and 3 respectively which are mainly attributed to νM-N and νM-O respectively. The low energy pyridine ring in plane and out of plane vibrations observed in the spectrum of the two ligands at 625 and 635 cm−1 respectively whereas the corresponding bands for the complexes are shifted to higher frequencies at 631–608 cm−1, 662–682 cm−1 and 690–714 cm−1 for 1, 2, and 3, respectively, which is a good indication of the coordination of the heterocyclic nitrogen [43].
UV–Vis spectra
UV–Vis spectra of 1, 2, and 3 were recorded at 300 K in HPLC grade acetonitrile solution. For 1 and 2 charge transfer bands were obtained at the wavelengths 378 and 365 nm. For 3, containing distorted ochtahedral Mn(II) ion, we obtained two bands in the UV region. The band obtained at 374 nm is considerably stronger than the band obtained at 247 nm. According to literature these bands are mainly assigned due to n→π* (forbidden transition) and π→π* transition in the ligand. It is well known that the d–d transition occur in d5 system but those transitions are of very low intensities, and hence, in 3 we did not observe any d–d bands for such transitions [44].
Cyclic voltammetry
Electrochemical property of 3 was studied in HPLC grade acetonitrile medium with tetrabutylammonium perchlorate as supporting electrolyte at a scan rate 50 mVs−1. The complex 3 shows an oxidative response at 0.51 V versus SCE, which is assigned to the Mn(II) to Mn(III) oxidation and a reductive response at 0.44 V versus SCE assigned for Mn(III) to Mn(II). This oxidation is reversible, characterized by a peak-to-peak separation (ΔEp) of 70 mV, which remain unchanged upon changing the scan rate. The anodic peak current (i pa) is almost equal to the cathodic peak current (i pc), as expected for a reversible electron transfer process.
Magnetic study
The Mn(II) complex, 3 as a solid exhibits room temperature magnetic susceptibility as expected for isolated d5 transition metal centers. The effective magnetic moment (μeff) value is found to be 5.92 B.M. at 300 K. The value is consistent with the magnetically dilute high spin d5 Mn(II) complex [45].
Description of the crystal structures of the complexes
[Zn(pbh)2]: (1)
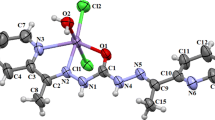
A perspective view of 1 with the atom numbering scheme is presented in Fig. 2 as an ORTEP plot (displacement ellipsoid are drawn at the 50% probability level for non-hydrogen atoms). Selected interatomic distances and angles are provided in Table 2. In 1 the Zn(II) center is coordinated by two deprotonated Hpbh ligand arranged in mer position. The large deviation from perfect octahedron of the six coordinated Zn(II) center in 1 has occurred due to the coordination of the polydentate N2O donor ligand (pbh) and as the chromophore is a heteroatomic species. The trans base angels [N2A–Zn–N2B and O1A–Zn–N1A] show marked deviation from their ideal value of 180°. All the Zn(II)–O bonds lie in the range from 2.2245(17) to 2.1145(18) Å whereas the Zn(II)–N bond lengths vary appreciably. One of the metal nitrogen bonds is very large [Zn–N1B = 2.315(2) Å] in comparison to the others [Zn–N2B = 2.057(2), Zn–N2A = 2.097(2) and Zn–N1A = 2.245(2) Å] and imply a highly distorted ochtahedral structure. The hydrazone ligand (Hpbh) coordinates in its enoleto form and thus acts as mono-negative ion. There is one strong intermolecular H-bonding interaction between the H4AA of one unit and O1B of the another unit of the complex. H-bonding diagram and H-bond dimension parameters are reported in Fig. 3 and Table 3 respectively.

ORTEP view of 1 with atom labeling scheme

Packing diagram of the complex 1
[Zn(acpbh)2]: (2)
A perspective view of 2 with the atom numbering scheme is presented in Fig. 4 as an ORTEP plot, and the packing diagram is shown in Fig. 5 (displacement ellipsoid are drawn at the 50% probability level for non-hydrogen atoms). Selected interatomic distances and angles are provided in Table 4. In 2, the angles at the Zn(II) center show small deviations from the ideal octahedral environment (indicated in Table 4). The dihedral angle between the two acpbh ligands is 149.96(3)°. In the ZnN4O2 coordination sphere of 2, Zn(1)–N(15) is the largest interatomic distance whereas the Zn(1)–N(9) is the shortest interatomic distance. In the complex, the three rings i.e. 2-acetylpyridine and the two five-membered chelate rings lie in the same plane. No classical H-bonding is present in the complex but an interlayer π–π interaction is present in 2 which is clearly shown in the packing diagram (Fig. 5).

ORTEP view of 2 with atom labeling scheme

Packing diagram of the complex 2
[Mn(acpbh)2]: (3)
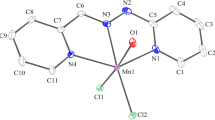
The ORTEP plot of 3 is illustrated in Fig. 6. Selected interatomic distances and angles are provided in Table 5. The Mn(II) ion is in N4O2 coordination sphere. Each of the two meridionally spanning ligands (Hacpbh) coordinates the metal ion via the pyridine–N, imine–N and deprotonated amide–O atoms forming two five membered chelate rings. The average N–N (1.385 Å), N–C (1.332 Å) and C–O (1.275 Å) distances in the {= N–N=C(O−)–} moiety of both ligands conform with the enolate form of the amide functionalities. A large distortion of the MnN4O2 octahedron is clearly evident from the bond distances and angles given in Table 5. The intraligand bite angles lie in the range of 91.13−93.48°. An interesting feature of the structure is that the two acpabh moieties are nonequivalent with respect to the metal to coordinating atom distances. The bond distances between the manganese and imine nitrogen atoms are 2.191 and 2.194 Å for Mn(1)–N(5) and Mn(1)–N(2) respectively. Moreover Mn(II) pyridine–N and Mn(II) amide–O distances for the two chelating ligands are significantly different. Average distance between Mn(1)–O(1) and Mn(1)–O(3) is 2.142 Å and between Mn(1)–N(1) and Mn(1)–N(4) is 2.294 Å Thus a tetragonal elongation is operative along the N(1)–Mn–N(4) axis at the same time a tetragonal compression exists along the O(1)–Mn–O(3) axis. Such compression and elongation results in a rhombic distortion from octahedral symmetry has been noted for similar tridentate ligands.

ORTEP view of 3 with atom labeling scheme
Fluorescence study of 1 and 2
The fluorescence spectra of 1 and 2 are presented in Figs. 7 and 8 respectively. The free ligands (Hpbh and Hacpbh) show intense emission bands at 330 and 340 nm, respectively, when exited with light of 378and 364 nm, respectively. These band intensities are reduced many fold in the case of 1 and 2 and blue shifted to 420 and 440 nm respectively. Quenching of fluorescence of ligands by transition metal ions upon complexation is a rather common phenomenon and can be explained by processes such as magnetic perturbation, redox activity, electronic energy transfer, donation of lone pairs of electrons, etc. [46, 47]. As the emission bands of the free ligands show reduction of intensities on complexation so it can be attributed to n–π* transition rather than π–π* transition [48]. As the lone pairs of electrons on the free ligands get donated to the metal centers in the complexes so the n–π* emission intensities get reduced in the complexes and explains the quenching of fluorescence in 1 and 2.

Normalised emission spectra of Hpbh (■) and 1 (□) at room temperature (λex = 378)

Normalised emission spectra of Hacpbh (■) and 2 (□) at room temperature (λex = 364)
EPR study of 3
The X-band EPR spectrum from a solution of [Mn(acpbh)2] (3) in DMSO at 10 K is shown in Fig. 9. The spectrum were recorded with the external magnetic field oriented either perpendicular or parallel to the microwave magnetic field. In perpendicular mode, signals are observed in a wide spectral region. A sextet of lines is observed at g ∼ 2.0 which is attributed to Mn(II) (S = 5/2) system. In parallel mode, a broad signal is observed with a valley at g ∼ 5.45. The spectrum are consistent with a Mn(II) (S = 5/2) monomer. The X band EPR signals of Fig. 9 show similarities with signals of other Mn(II) (S = 5/2) monomers [49–54]. Qualitatively, the spectrum can be interpreted assuming a zero field splitting parameter, |D| of the order of 0.1 cm−1. Under these conditions, the signals observed in parallel mode arise from inter-doublet transitions. The lack of resolved hyperfine patterns, due to the I = 5/2 55Mn nucleus, is attributed to a distribution on the zero field splitting parameters [54]. A more detailed analysis including high field/high frequency EPR studies, will be presented elsewhere.

X-band dual mode EPR spectra from a DMSO solution of 3. EPR conditions: temperature, 10 K, microwave frequency, 9.60 GHz (B1⊥B), 9.32 GHz (B1‖ B), microwave power, 2.1 mW, modulation amplitude 10 Gpp
Conclusion
The two different N,N,O donor hydrazone ligands (Hpbh and Hacpbh) have efficiently coordinated to Zn(II) and Mn(II) ions producing similar structures. Inspite of varying the carbonyl group attached to the pyridine moiety present in the hydrazone ligand in both the Schiff bases, we obtained three mononuclear complexes of the type [Zn(pbh)2] (1), [Zn(acpbh)2] (2) and [Mn(acpbh)2] (3) which indicate that the alkyl group does not show any pronounced effect on the ligating properties of the two ligands.
Supplementary data
X-ray crystallographic data in the ‘CIF’ format corresponding to the complexes reported in this article have been deposited with the Cambridge crystallographic Data Center and supplementary crystallographic data for this paper can be obtained free of charge on request at http://www.ccdc.cam.ac.uk/conts/retrieving.html [or from the Cambridge Crystallographic Data Centre (CCDC), 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44(0)1223-336033; email: deposit@ccdc.cam.ac.uk], quoting the CCDC numbers 648266, 648735 and 632494 for the complexes 1, 2 and 3 respectively.
References
(a) Craliz JC, Rub JC, Willis D, Edger J (1955) Nature 34:176, (b) Iskander MF, Zayan SE, Khalifa MA, El-Sayed L (1974) J Inorg Nucl Chem 36:556
(a) Merchant JR, Clothia DS (1970) J Med Chem 13:335, (b) Dilworth JR (1976) Coord Chem Rev 21:29
Ranford JD, Vittal JJ, Wang YuM (1998) Inorg Chem 37:1226
Wester D, Palenik GJ (1976) Inorg Chem 15:755
Andjelkovic K, Ivanovic I, Prelesnik B, Leovac VM, Poketi D (1996) Polyhedron 15:4361
Abram S, Maichle-Mössmer C, Abram U (1998) Polyhedron 17:131
Carcelli M, Ianelli S, Pelagatti P, Pelizzi G (1999) Inorg Chim Acta 292:121
Zhou J, Chen Z-F, Wang X-W, Tan Y-S, Liang H, Zhang Y (2004) Acta Crystallogr E60:m568
Ali H, Khamis NA, Basirun WJ, Yamin BM (2004) Acta Crystallogr E60:m1351
Mohamed GG, El-Wahab ZHA (2003) J Thermal Anal Calorimetry 73:347
Song Y, Xu Y, Zhu D-R, You X-Z (2000) Acta Crystallogr C56:430
Seth S, Chakraborty S (1984) Acta Crystallogr C40:1530
de Sousa GF, Filgueiras CAL, Abras A, Al-Juaid SS, Hitchcock PB, Nixon JF (1994) Inorg Chim Acta 218:139
Andjelković K, Ivanović I, Niketić SR, Prelesnik B, Leovac VM (1997) Polyhedron 16:4221
And-jelković K, Jakovljević G, Zlatović M, Tešić Ž, Sladić D, Howing J, Tellgren R (2004) J Serb Chem Soc 69:651
Bonardi A, Merlo C, Pelizzi C, Pelizzi G, Tarasconi P, Vitali F, Cavatorta F (1991) J Chem Soc, Dalton Trans 1063
Palenik GJ, Wester DW (1978) Inorg Chem 17:864
(a) Rees DC, Lewis M, Lipscomb WN (1983) J Mol Biol 168:367, (b) Holland DR, Haustrath AC, Juers D, Matthews BW (1995) Protein Sci 4:1955
(a) Nair SK, Christianso DW (1991) J Am Chem Soc 113:9455, (b) Silverman DN, Lindskog S (1988) Acc Chem Res 21:30
Kim EE, Wyckoff HW (1991) J Mol Biol 218:449
Weighardt K (1989) Angew Chem Int Ed Engl 28:1153
(a) Yachandra VK (1996) Chem Rev 96:2927, (b) Yagi M, Kaneko M (2001) Chem Rev 101:21
Dismukes GC (1996) Chem Rev 96:2909
Pecoraro VL (ed) (1992) Manganese redox enzymes. VCH publishers, New York
(a) Kono Y, Fridovich I (1983) J Biol Chem 258:6015, (b) Beyer WF Jr, Fridovich I (1985) Biochemistry 24:6460
Barynin VV, Hempstead PD, Vagin AA, Antonyuk SV, Melik-Adamyan WR, Lamzin VS, Harrison PM, Artymyuk PJ (1997) J Inorg Biochem 67:196
Allgood GS, Perry JJ (1986) J Bacteriol 168:563
Whittakar JW, Sigel A, Sigel H (eds) (2000) Metal ions in biological systems, vol 37. Marcel Dekker, NewYork, 505 pp
Tommos C, Babcock GT (1998) Acc Chem Res 31:18
Zouni A, Witt HT, Kern J, Formme P, Kraub N, Saenger W, Orth P (2001) Nature 409:739
Dinda R, Sengupta P, Ghosh S, Sheldrick WS (2003) Eur J Inorg Chem 2:363
Dinda R, Sengupta P, Ghosh S, Figge HM, Sheldrick WS (2002) Dalton Trans 4434
Gohdes JW, Armstrong WA (1992) Inorg Chem 13:368
Sheldrick GM (1999) SHELXTL Programs for the solution and refinement of crystal structures: ver. 612, Bruker AXS P4 Inc., Madison
Sheldrick GM (1996) SADABS, Program for area-detector absorption correction: University of Göttingen, Germany
CrysAlis PRO (2007) Crysalis Software system: Oxford Diffraction Ltd, Abingdon, UK
SMART and SAINT-PLUS (2003) Area Detector Control and Integration Software: ver. 5629 and 645, Bruker Analytical X-Ray Instruments, Inc., Madison
Spek AL (2003) J Appl Crystallogr 36:7
Sheldrick GM (1990) SHELXS-97, Program for crystal structure refinement. Universityof Göttingen, Gottingen
Sheldrick GM (1997) SHELXL-97, Program for crystal structure refinement. University of Gottingen, Gottingen
Addison AW, Rao TN, Reedijk J, van Rijn J, Verschoor GC (1984) J Chem Soc, Dalton Trans 1349
Conley RT (1966) Infrared spectroscopy. Allyn & Bacon, Boston
Nakamoto K (1997) Infrared and Raman spectra of inorganic and coordination compounds, vol 23. Wiely, New York
Lever ABP (1984) Inorganic electronic spectroscopy. Elsevier Science, New York
Batra G, Mathur P (1994) Transit Met Chem 19:160
Varnes AW, Dodson RB, Wehry EL (1972) J Am Chem Soc 94:94
Kemlo JA, Sheperd TM (1977) Chem Phys Lett 47:158
Proc. Indian Acad. Science (Chem Science) (2001) vol 113, 285 pp
Goodgame M, Hayward PJ (1971) J Chem Soc A:3406
Birdy RB, Goodgame M (1979) Inorg Chem 18:472
Goodgame M, Hussain I (1995) Inorg Chim Acta 229:165
Whittaker MM, Whittaker JW (1997) Biochemistry 36:8923
Goodgame DML, Mkami HE, Smith GM, Zhao JP, McInnes EJL (2003) Dalton Trans 34
Pierce BS, Elgren TE, Hendrich MP (2003) J Am Chem Soc 125:8748
Acknowledgments
We sincerely acknowledge Department of Science and Technology, New Delhi, Govt. of India, for the financial support. We also acknowledge Dr. Andres Ibañez of University of Chile and Dr. Andrés Goeta of Durham University for performing the structural measurement of complex 3.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ray, A., Banerjee, S., Sen, S. et al. Two Zn(II) and one Mn(II) complexes using two different hydrazone ligands: spectroscopic studies and structural aspects. Struct Chem 19, 209–217 (2008). https://doi.org/10.1007/s11224-007-9274-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-007-9274-7