
Abstract
New Schiff base; (S)-4-(methylthio)-2-((1-(thiophen-2-yl)ethylidene)amino) butanoic acid (LH); was synthesized by condensation of 2-acetyl thiophene with methionine in a mole ratio of 1:1 and characterized with1H-13C NMR, UV–Vis, FT-IR and MS methods. The metal complexes of manganese(II), cobalt(II), nickel(II), copper(II), and zinc(II) were prepared by direct reaction of the ligand (LH) and the metal chlorides which structurally characterized with spectroscopic methods, magnetic, and conductivity measurements. The data showed that the Schiff base LH ligand behaves as a bidentate via nitrogen atom of imine group and oxygen atom of carboxylic methionine moiety. The results obtained from magnetic moments, electronic spectra, mass spectra and elemental analyses confirmed the tetrahedral geometry around the central metal ions. The optimization of geometry, vibrational band assignments, frontier molecular orbitals, and absorption spectrum of the compounds were investigated by DFT/B3LYP/6–31 + G(d,p) methods. Natural bond orbital analysis was carried out for the investigation of major stabilizing orbital interactions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Recent years witness a growing interest in the chemistry of transition metal complexes involved amino acids moiety due to serving biological systems [1, 2]. The derivatives of thiophene are widely distributed in the nature and have a variety of biological applications [3, 4]. The coordination of the nitrogen atom of azomethine group to a metal ion lead to induction of acidic property of the ligand, which have resulted in a more active in their based-ligand of amino acids [5, 6]. Consequently, the hydrogen atoms of the azomethine group are able to form intermolecular hydrogen-bonds with the carboxylate oxygen of the amino acid [7–10].
The transition metal complexes of poly dentate ligands with methionine amino acid have received a great attention in spite of their potential metal binding properties and promising applicability [8, 9]. These Schiff base ligands provided the synthetic models for the metal-binding sites on the proteins and enzymes [10, 11]. Hence, the synthesized Schiff bases serve as an important intermediate in many enzymatic reactions involving the interaction of enzyme with amino or carbonyl groups [12, 13].
The preparations of template complexes involving heterocyclic rings like thiophene and amino acids have been investigated to evaluate antimicrobial properties of these compounds [14]. The coordination chemistry of l-methionine Schiff bases contributed to constitute a new type of potential antibacterial, anti-inflammatory, and anticancer agent [15, 16]. In particular, the Schiff base derivatives of thiophene show a dramatic increase of the diversity of biological properties in recent years [17].
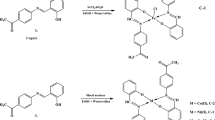
The present work deals with the synthesis and characterization of a new Schiff base, which was prepared via condensation of 2-acetyl thiophene with methionine to produce (S)-4-(methylthio)-2-((1-(thiophen-2-yl)ethylidene)amino) butanoic acid Schiff base ligand (LH) and its complexes (M(II)-L) with manganese(II), cobalt(II), nickel(II), copper(II), and zinc (II) (see Scheme 1).

Synthesis of LH Schiff base by reaction of methionine with 2-acetyl thiophene
Density-functional theory (DFT) has been widely applied to determine the structural and spectral parameters of systems involving transition metal complexes, which offered an alternative method for the quantum mechanical computation of chemical properties [14, 18]. Hence, the goal of the work is to prepare the new Shiff base of methionine amino acid, LH and its transition metal complexes, including Mn(II)-L, Co(II)-L, Ni(II)-L, Cu(II)-L, and Zn(II)-L and also to evaluate the electronic structure of the title compounds using density functional theory (DFT) calculations at B3LYP/6–31 + G(d,p) level of theory in the gas phase.
The frequency calculations were performed on all the optimized geometries to elucidate the IR spectra and also to ensure that the obtained structures represent local minima. The NMR shielding tensors for LH ligand were calculated with B3LYP method and 6–31 + G(d,p) basis set in DMSO media. Time-dependent density functional theory (TD-DFT) is a powerful tool allowing for accurate description of excited electronic states in transition metal complexes [19]. The excited states of the electronic transition probabilities of the representative transition metal complexes were investigated in the framework TDDFT/PCM procedures. In addition, the natural charges of the compounds and the energy gap between the frontier orbitals were studied by natural bond analysis NBO method [20].
Experimental
Physical methods
All the chemicals used were either of AR or chemically pure grade. The elemental analysis was carried out using a Heraus-CHN-rapid analyzer. Metal contents were estimated using an AAS Shimadzu 670 spectrometer at AL-Mustansiriya University laboratories. The 13C and 1H NMR spectra were recorded on a Bruker NMR spectrometer 400 MHz in DMSO d6 at laboratories of Cairo University (Egypt). The FTIR spectra were recorded in the 4000–400 cm−1 region on a Shimadzu IR-435 in KBR and CsI discs. The UV–Visible spectra of the title complexes and free Schiff base LH in methanol and DMF solutions were recorded on a Cary-2390 UV–Vis spectrophotometer. The magnetic susceptibilities were measured on Sherewood Auto Magnetic balance at room temperature using Hg[Co(NCS)4] as calibrate. The mass spectra were recorded on a GCMS_PQ 2010 Ultra Shimadzu instrument. The molar conductance measurements were obtained using 10−3 molar solutions in DMF with a WWT. Conductivity meter bridge (Model CM-180) and dip type cell calibrated with KCl solutions.
Synthesis of (S)-4-(methylthio)-2-((1-(thiophen-yl)ethylidene)amino)butanoic acid (LH)
The new Schiff base (LH) was prepared by mixing of an ethanolic solution (25 mL) of methionine (0.01 mol, 1.49 g) with 2-acetyl thiophene (0.01 mol, 1.27 g) and then refluxing the mixture on the water bath for 2 h. After concentration of the solution, the yellow precipitate was separated, filtered, washed with ethanol and chloroform, and then dried over anhydrous calcium chloride. The recrystallization from hot methanol to afford pale yellow crystals (see Scheme 1). Chem. Anal. Calc. for C11H15NO2S2 (MW: 257.37): C, 51.33; H, 5.87; N, 5.44; S 24.92%. Found: C, 50.99; H, 5.00; N, 4.71; S, 23.7%. M.P. (180–182 °C). IR ν (KBr disc)/cm−1: 3500–2800 (br), 2960(m), 1678(s), 1630 (s), 800 (w), 650(w). Molar conductance Λm/Ω−1 cm2 mol−1 (in DMF):10
Synthesis of the metal complexes of Mn(II), Co(II), Ni(II), Cu(II), and Zn (II): general procedure
A mixture of LH metal chloride (10 mmol, 0.257 g) in methanol (10 mL) was added drop wise to an aqueous solution (10 mL) of hydrate metal chloride (10 mmol) of manganese (II), cobalt (II), nickel (II), copper (II), and zinc (II). The mixture of reaction was refluxed for 3–4 h in water bath and then excess solvent was removed by vaporization under reduced pressure. The colored precipitate compounds that separated were filtered, washed with distilled water, ethanol, and dried over anhydrous calcium chloride (Scheme 2).

Synthesis of metal complexes of Schiff base (M(II) = Mn, Co, Ni, Cu, Zn)
[MnL(H2O)Cl]
Chem. Anal. Calc. for C11H16NO3S2ClMn (MW: 364.72): Mn, 15.06; C, 36.22; H, 4.42; N, 3.84; S 17.58%. Found: Mn, 13.90% C, 35.91; H, 4.98; N, 4.01; S 18.00%. M.P (295 °C). IR ν (CsCl disc)/cm−1: 2963 (m), 1670 (s), 1610 (sh), 810 (w), 640(w), 520 (m). Λm/Ω−1 cm2 mol−1 (in DMF): 44. Color: yellow.
[CoL(H2O)Cl]
C11H16NO3S2ClCo (MW: 368.77): C, 35.83; H, 4.37; N, 3.80; S, 17.39; Co, 15.98%. Found: C, 34.40; H, 4.07; N, 3.07; S, 16.66; Co, 16.00%. M.P: (300 °C). IR ν (CsCl disc)/cm−1: 2966 (m), 1667 (s), 1605 (sh), 968 (w), 640 (w), 500 (w), 400 (m). Λm/Ω−1 cm2 mol−1 (in DMF): 44. Color: green.
[NiL(H2O)Cl]
C11H16NO3S2ClNi (MW: 368.53): Ni, 15.93; C, 35.85; H, 4.38; N, 3.80; S, 17.40%; Found: C, 34.40; H, 3.93; N, 3.04; S, 16.11; Ni, 15.80%. M.P: (278 °C). IR ν (CsCl disc)/cm−1: 2962 (m), 1660 (s), 1580 (sh), 760 (w), 650 (w), 480 (w), 430 (m). Λm/Ω−1 cm2 mol−1 (in DMF): 50. Color: brown.
[CuL(H2O)Cl]
C11H16NO3S2ClCu (MW: 373.38): Cu, 17.02; C, 35.38; H, 4.32; N, 3.75; S, 17.17%. Found: Cu, 16.00; C, 34.35; H, 3.66; N, 3.99; S, 16.33%. M.P: (277 °C). IR ν (CsCl disc)/cm−1: 2950 (m), 1655(s), 1566 (sh), 810 (w), 643 (w), 540 (w), 420 (m). Λm/Ω−1 cm2 mol−1 (in DMF): 69. Color: beige.
[ZnL(H2O)Cl]
C11H16NO3S2ClZn (MW: 375.22): Zn, 17.43; C, 35.21; H, 4.30; N, 3.73; S, 17.09%. Found: Zn, 17.00; C, 34.00; H, 3.29; N, 4.61; S, 16.07%.. M.P: (301 °C). IR ν (CsCl disc)/cm−1: 2963 (m), 1658 (s), 1560 (sh), 760 (w), 648 (w), 508 (w), 424 (m). Λm/Ω−1 cm2 mol−1 (in DMF): 47. Color: off white.
The computational details
All computations were performed by density functional theory (DFT) in the framework of the Becke three-parameter hybrid exchange and Lee–Yang–Parr correlation functional (B3LYP) using GAUSSIAN09 program suite [21]. The basis sets were described with a split-valenc Pople basis set plus polarization and diffuse functions, 6–31 + G (d,p) for H, C, N, O, and S atoms, but a double-ζ quality LANL2DZ basis set was employed for transitional metal atoms [22]. All the “inner electrons” of metal ions were replaced and described with a scalar relativistic electron core potential (ECP). Frequency calculations were performed on all optimized geometries to ensure that the obtained structures represent local minima. The calculated vibrational frequencies were obtained with the use of a scaling factor of 0.9614 as recommended for the B3LYP/6–31 + G(d,p) level [23]. The 1H NMR and 13C NMR chemical shifts are calculated within GIAO approach [24], which is one of the most common approaches for calculating nuclear magnetic shielding tensors. Natural bond analysis (NBO) was applied to analyze the electronic properties and charge transfer of the complexes [20]. To determine the absorption spectra, the ground-state structure of all the complexes were first optimized in the solvent media. Then, the vibrational frequencies were evaluated, to check the absence of imaginary frequencies. Finally, the absorption spectrum, in the singlet excited state, has been obtained for the titled compounds using time-dependent density functional theory (TD-DFT) calculations at B3LYP/6–31 + G(d,p) level with the polarized continuum model (PCM) in methanol solvent [25].
Results and discussion
The new Schiff base, LH derived from condensation of equimolar ratios of a methionine amino acid with 2-acetyl thiophene have used as chelating agent to form new metal complexes with Mn(II), Co(II), Ni(II), Cu(II), and zinc(II) ions. All the complexes were sparingly soluble in common organic solvents such as ethanol, methanol, and chloroform, whereas they were soluble in DMF, DMSO, and acetonitrile. The analytical data indicated that all the complexes are mononuclear in nature, while the E-configuration is most stable with respect to other configurations since it is favored one mole in coordination with central metal. The molar conductance values measured in DMF solution (1 × 10−3 mol dm−3) fall in the range 29–33 Ω−1 cm2 mol−1 supporting their nature behavior in solution due to absence of anionic chloride counter ions in the outer sphere structure [14].
The 1H NMR analysis
Further evidence for the formation of LH Schiff base was obtained from 1H NMR spectra (Figure S1), which provides diagnostic tool for the positional elucidation of the protons. The protons belonging to the aromatic thiophene moiety were observed within the expected chemical shift [26] in the region 6.10–7.90 ppm as multiple absorption. Also, the acidic proton of the carboxylic –COOH resonance was displayed at 10.10 ppm as singlet peak [8, 10]. However, the shielded protons of –CH2– and CH3–S moiety were resonated in the regions around 1.415–1.449 and 2.31–2.44 ppm, as multiple peaks suggesting the condensation of the methionine amino acid with the carbonyl group of 2-acetyl thiophene. Also, the broad singlet peak at 3.45 ppm is due to CH2–S of methionine fragment and CH3 group attached to N=C. The singlet peak at 4.43 ppm could be assigned to H–C=N– moiety due to the electronic environment of imine and carboxylic groups [9, 10, 14].
The 13C NMR spectra of LH ligand showed at least 10 distinguishable carbons by supporting different 13C chemical shifts (Figure S2). The C=O carboxylic acid signal showed down filed chemical shift at 188.9 ppm. The carbon peaks belonging to the thiophene ring were recorded at 117–136 ppm and the C=N signal were appeared at 169.6 ppm [27]. The signals of methyl and methylene carbons were observed at 15.9–21.3 ppm except the shielded aliphatic proton of HC–N observed at 74.8 ppm.
The mass spectra
The mass spectra of the Schiff base (LH) and its manganese(II) complex are shown in Fig. 1a, b, respectively. Figure 1 (a) exhibits the molecular ion M1+ for the Schiff base of m/e = 258, which agrees well with the suggested formula of the prepared ligand. Also, the other peaks at 243 (base peak), 213, 197, 156, and 102 are attributed to cleavage of the methyl, carboxylic, and other fragments of methylene moiety of methionine amino acid [28]. However, Fig. 1b shows the molecular ion at 365 and 367 assigned to M+ and M+2 for [MnL(H2O)Cl] complex due to isotopic of sulfur present in the structure of the organic moiety. The fragments at 333, 332, and 285 are assigned to cleavage of Cl (333, 332M-Cl) with effect of chlorine isotopes and cleavage of –S–CH3 moiety respectively. The results obtained from mass spectra give good proof to elucidate the suggested structures of the prepared compounds.

a The mass spectra of LH Schiff base, b [MnL(H2O)Cl] complex
IR spectra
The fundamental vibrational bands of the ligand LH, as well as its metal complexes, were investigated by IR spectroscopy (shown in Figure S3). The free ligand showed a broad band at 3500–2800 cm−1 assignable to the –OH moiety and two intense bands at 1678 and 1630 cm−1 corresponding to the –C=O and –C=N stretching of the carboxylic group. The comparison of the positions of these bands with those observed in the infrared spectra of its Mn(II), Ni(II), Co(II), Cu(II), and Zn(II) complexes indicated that after complexation, the broad band nearly disappeared after coordination of O atom of carboxylate group along with complexation, indicating involvement of COOH in the coordination with the metal ions via deprotonation [29]. Furthermore, the absorption of –C=O regarded to –COOH is subjected to red shift in the region 1655–1670 cm−1 in metal complexes. Furthermore, the lowering in the wave numbers of imine –C=N– moiety in the infrared spectra of complexes supported the formation of M–N bond [30]. Also, a strong intensity band at 1630 cm−1 assigned to ʋ–C=N isomethine vibrations [30, 31] was shifted to lower wavenumbers at region of 1610–1560 cm−1 in the complexes, supporting the formation of M–N bonds. The weak to medium intensity bands at 2962–3010 cm−1 and 650-810 cm−1 are attributed to aliphatic C–H and C–S bonds [10, 28, 31]. However, the weak intensity vibrations in the regions of 400–450 and 500–540 cm−1 revealed the M–N and M–O coordination bonds which are formed up on chelating of the metal ions with the bidentatate monobasic Schiff base LH [32]. Unfortunately, the identification of M–Cl was not possible due to processing the FTIR spectra in the range 4000–400 cm−1.
The UV–visible spectra and magnetic moments
The solution of LH Schiff base in methanol displayed two peaks at 498 and 274 nm, which are ascribed to π → π* and intra-ligand charge transfer of the chromospheres moiety involved in the structure of the ligand [7, 33]. The UV–Vis spectra of Schiff base ligand LH and also Ni(II) and Co(II) complexes were shown in (Figures S4 and S5). The pink solution of Mn(II) complex in DMF exhibited one spin-forbidden transition at 22,222 cm−1 (450 nm) of d–d regarded to tetrahedral geometry [23]. The cobalt (II) complex showed a weak energy bands and near infra-red region at 11,284 and 11,600 cm−1 indicating the tetrahedral geometry [34]. Also, the high-spin state for manganese (II) and cobalt (II) complexes via their paramagnetic properties at 5.90 and 4.00 BM are in agreement with the proposed geometry [25]. The electronic spectra of nickel(II) and copper(II) complexes showed spin-allowed transitions in the regions 17,094–18,281 and 14,492 cm−1 assigned to 3T1 → 3T2, 3T1 → 3T1(p) (Ni2+) and 2T2 → 2E (Cu2+). The values obtained of magnetic moments for Ni(II) and Cu(II) complexes at room temperature are 2.88 B.M and 1.20 B.M, respectively, corresponding to their expected values of tetrahedral geometry [34]. Since no d–d transitions or magnetic moment occurs for Zn(II)-Schiff base, the environment around Zn(II) with LH, acting as a bulky bidentate ligand, is considered a tetrahedral geometry [35]. Thus, the magnetic moment measurements helped us to predict the possible geometry of the prepared metal complexes.
Computational approaches
Geometry optimization
The electronic structures of the Schiff base (LH) and also the metal complexes, Mn(II)-L, Co(II)-L, Ni(II)-L, and Zn(II)-L, were optimized using DFT calculations at the B3LYP level of theory without any restrictions on the internal coordinates in the gas phase. All the stationary points were characterized by their harmonic vibrational frequencies as minima or saddle points. Figure 2 shows the optimized structures and atom labeling via some geometrical parameters (bond lengths and angles) of above mentioned compounds. The optimization calculations for the Schiff base, LH, and M(II)-L complexes are discussed in four parts:


The optimized structures and atoms labling of a Shiff base ligand (LH), b Mn(II)-L, c Co(II)-L, d Ni (II)-L, and e Zn(II)-L complexes, showing with some bond lengths and bonding angles
The bond length of –C=O of carboxylic moiety is 1.208 Å, which is shifted to longer bonds with respect to the metal complexes (1.246–1.244 Å), assigning to deprotonation the carboxylic group on coordination with the metal ions. Also, the bond length of imine –C=N– group is 1.286 Å in the free LH ligand, whereas the longer bond length were observed in all the complexes in the range of 1.309–1.445 Å. These observations can be attributed to the movement of lone-pair of nitrogen towards vacant orbitals of metal ions with high strength bonding of M–N, i.e., the Cu–N with 1.995 Å in comparison with Mn–N of 2.216 Å. Since the Mn(II) tends to form less stable complexes in regarding to Irving–Williamson series.
The unremarkable changes in some bonds lengths such as –C=C, –C–S–CH3, –C=CH–S, and –C–O might be attributed to impossibility to participation in bonding with the metal ion. The increasing M–(O–C=O) bond length from Cu–(O–C=O) to Mn–(O–C=O) results from the covalence character in bonding of Cu(II) ion with negatively charged carboxylate group.
However, M–Cl geometry parameters exhibit good evidences for the coordination of chloride ion to central metal ion with bond lengths 2.277–2.341 Å and bond angles ∠ClMN = 107.6–146.8 and ∠ClMO1 = 88.5–111.0 Å, which can assert a tetrahedral geometry for all metal complexes. The presence of coordinated water molecules in the inner sphere of metal complexes [MLCl(H2O)] can be investigated from the M–O bond length in the range of 1.995–2.151 Å.
The relative stabilization energies with respect to LH ligand were calculated as follow: Cu ≫ Ni > Co > Mn > Zn. This result indicates that the Cu(II) complex tends to form more stable complexes (about twice) of such Schiff base ligands than the other transition metals.
Frequency calculations
In order to resolve the fundamental absorption spectra, the IR spectra corresponding to LH Schiff base and above-mentioned complexes were characterized using vibrational frequency analysis. The calculated IR spectra for LH ligand (upper bands) and its complexes (lower bands) with the assignment of their fundamental vibrational modes are presented in Fig. 3. A strong sharp band was observed at 3512 cm−1 due to νOH of carboxylic group present to LH Schiff base (exp: 3500–2800 cm−1), which disappeared in the metal complexes [14]. The band corresponds to–C=O group of LH ligand was red shifted from 1872 to 1696 cm−1 in the metal complexes (exp: 1678 to 1655–1670 cm−1). The C=N vibrations were observed as a sharp band at 1696 cm−1 (LH ligand) and 1624 cm−1 (metal complexes) [27]. A few weak to medium peaks at 2992–3216 and 660–820 cm−1 (exp: 2962–3010 and 3650–810 cm−1) are attributed to aliphatic C-H and C–S bonds. The aromatic CH stretching vibrations show a few signal at about 3176 cm−1 [36]. The weak to medium absorptions in the regions 400–450 and 500–607 revealed the M–N and M–O coordination modes which were not found in free ligand [27, 29, 34].

The computed IR spectra for LH ligand (upper curve) and its metal complexes (lower curves) in the gas phase. Intensities are normalized to 1
NMR calculations
The isotropic chemical shifts using NMR analysis is well known as one of the reliable and powerful tools to predict and interpret the structure of organic and inorganic biomolecules [37]. In order to provide an unambiguous assignment of 13C and 1H NMR spectra of synthesized ligand LH, the theoretical 1H and 13C NMR chemical shifts of LH were calculated using GIAO approximation, and the obtained results were compared with experimental ones (Table 1). The PCM model for DMSO was also employed to describe the influence solvent on the NMR spectra. By considering to the atom labeling in the Fig. 4, the calculated 1H NMR spectra are summarized as follow: the carboxylic acid proton signal (H1) appeared at 12.46 ppm due to the presence of electron with-drawing carboxylate group [19]. The aromatic thiophene protons of attached to H2-H4 give overlapped signals with chemical shifts of 8.15 ppm, but the peak at 8.09 ppm is assigned to aromatic proton of thiophene [27]. The aliphatic proton signal of H5 appeared at 5.13 ppm due to higher electronegativity properties of imine and carboxylic groups. It was concluded that all the aliphatic protons attached to methyl (H8, H9, H10, H13, H14, and H15) and methylene (H6, H7, H11. and H12) are nonequivalent and give signals in the range of 3.42–2.55 ppm. In our calculations, the aromatic carbon signals (C8–C11) are calculated at 112–137 ppm [27]. The chemical shift value of the carboxylate carbon (C5) in the highest shift is assigned to 158 ppm due to the anisotropy effects of carbonyl group. The peak at 54 ppm is assigned to aliphatic C4 due to the effect of imine and carboxylate moiety. The methylene aliphatic carbon signals (C2 and C3) are observed at 25.3 and 25.8 ppm while the methyl carbon peaks (C1 and C7) appeared at high field shift 10.5 and 6.7 ppm, respectively.

The theoretical geometric structure of LH ligand, showing the atom-numbering scheme
Natural bond orbitals
The electron configurations of the metal ions as well as the natural charge of the some fragments of the ligand and central metal of the compounds were calculated using the natural population analysis method (NPA) in the gas phase and the results are summarized in Table 2. The variation of the natural charge on central metals is correlated with the natural population of the metal ions 3d orbitals [38]. Whereas the metal ion 3d population considerably increases from Mn(II) to Zn(II) (5.20–9.99), the 4s (0.21–0.32) and 4p (0.33–0.42) population insignificantly vary in all complexes. This trend indicates that the participation of the dσ orbitals in the coordination bond dominates the charge variation on the metal ions, i.e. with increasing 3d population of the all metal ions, except Zn(II), with a less positive the M(II) natural charge, subsequently the trend of X/Y to M(II) charge transfer increases [38, 39]. As seen, the ligand to metal charge transfer decreases in series: Cu(II) > Mn(II) > Ni(II) ≈ Co(II) > Zn(II). It seems that increasing charge transfer from COO− and Cl− ligands to metal ions leads to the weaker interaction between metals and the other ligands (H2O and N(=C)) and result in a more positive charge on other mentioned ligands [37]. As expected, the covalency of the M–L bond is ranked as follows: M–COO > M–Cl > M–N > M–OH2. As a result, the NPA results show that the M(II) 3d orbitals play a significant role in the valence shell of the complexes.
The metal–ligand interactions are investigated by the second-order perturbative analysis (SOPA) of the ligand LP (donor) and metal LP* (acceptor) orbitals. The SOPA results are presented as a list of second-order interaction energies (E 2) between donor and acceptor orbitals [38, 40].
To understand better the metal–ligand interactions and also to determine the ligand-to-metal charge transfer transitions in the coordination complexes, the second-order perturbation analysis (SOPA) of a Fock matrix was investigated in NBO Basis. The SOPA is introduced as a list of second-order interaction energies (E2) between donor and acceptor orbitals of the ligand LP (donor) and metal LP* (acceptor) orbitals. These non-covalence interactions are estimated by the second-order correction to interaction energy, E 2, between the occupied molecular orbitals of Lewis-type NBOs (donor) and the neighboring unoccupied molecular orbitals non-Lewis NBOs (acceptor). The interaction energy E 2, associated with delocalization (2e−stabilization) i → j is estimated by Eq. (1) [35]:
where i and j are donor and acceptor NBOs, respectively, and q i is the donor orbital occupancy, εi, εj are diagonal elements (orbital energies), and F(i,j) is the off-diagonal NBO Fock matrix element.
Using the second-order perturbation analysis, we can obtain a qualitative picture of interactions between the central metal orbitals (3d, 4s, 4p) and the ligands. Whereas the E 2 energy of LP X/LP* M(II) is identified with the extent charge transfer from a lone pair electrons of ligand X to an antibonding NBO orbitals of metal atom (X → M), the E2 energy of M(II) LP/X LP* represents the charge transfer from metal to ligands (electron back-donation). The interaction energies, E(2), larger than 15.0 kcal mol−1 are summarized in Table 3, along with the contribution of the M(II) (3d, 4s, 4p) orbitals and the spn (n = 0.54–0.95) hybridized NBO orbitals of ligand groups. The atom labeling (vide infra) was shown in Fig. 2. The E2 energies of metal to ligand in all the complexes were very low and so are not reported. In Mn(II) complex, there are more charge transfer from Cl ligand and also O atoms of H2O and COO ligand orbitals to Mn valance orbitals with various contributions. The lone pair on Cl atom with occupancy of 0.91780 has a strong interaction with 4 s orbital of Mn with occupancies of 0.11386 by 32.2 kcal mol−1 of interaction energy. Therefore, covalency of the Mn-X bond is ranked as follows based on the calculated E2 energies: Cl− > OH2 ≈ COO− > N (=C). In Co(II) complex, a major charge transfer takes place through the lone pair electrons of Cl with occupancies of 0.90399 by 47.7 kcal mol−1 of interaction energy. It seems that the importance of the Mn 4s orbital in some of the Mn-X interactions is more than those for 4p and 3d orbitals while in Co(II), the 3d contribution in Co(II)-X interaction is more important than the other orbitals. Also Ni(II)–Cl and Zn(II)–Cl SOPA results are in same direction with Mn(II–Cl. On the other hand, Cu–N(=C) has a little more interaction energy than that of Cu–Cl, thus the Cu 4s orbital dominates the Cu–N interaction, whereas the Cu 4p orbital dominates the Cu–Cl interaction. Among these complexes, except Co(II) complex, the contribution from the 3d orbitals remains negligible in all cases. In summary, the calculated results demonstrate that the participation of the metal ions 3d orbitals, except Co(II), will be predominant in a less covalent metal–ligand bond whereas their 4s and 4p orbitals will play a more significant role in a covalent environment.
The frontier orbitals and TD-DFT calculations
The electronic absorption is defined as a process that involves transition of an electron from the ground state to the first excited state. The transitions occur from the highest occupied molecular orbital (HOMO; π donor) to the lowest unoccupied molecular orbital (LUMOl; π acceptor) [41]. The energy gap between HOMO and LUMO can be related to chemical activity of the molecules and tendency to intra-molecular charge transfer (ICT). The larger the HOMO–LUMO energy gap, the harder and more stable/less reactive the molecule [41].
In this study, HOMO and LUMO energies of LH and its complexes were calculated using DFT levels. Figure 5 shows the energy gap between the HOMO and LUMO orbitals in all the compounds. A small HOMO–LUMO bond gap (E p) favors electron hopping between both groups of occupied and unoccupied molecular orbital that enhance the charge transfer [19]. The HOMO (electropositive density) and LUMO (electronegative density) displayed via the green and red in each complex, respectively. The HOMO orbitals were localized mainly on Me-thio (99%) in all the compounds while LUMOs are vary distributed on the molecules. In Mn and Co and Zn complexes, LUMO orbitals were located mainly on thiophene and C=N groups (Mn: 45 and 43%, both spin state), Co (49 and 46% α-spin), and Zn (46 and 49%), but in β-spin Co, that is localized mainly on Co atom (45%), C=N (25%) and thiophene (23%). In α-spin Ni complex, the LUMO is distributed on thiophene (48%), C=N (29%) and Me-thio (21%) while in β-spin is located mainly on Ni atom (86%). In α-spin Cu, those are mainly on thiophene (49%) and C=N (45%) groups, but in β-state, the electron density are contributed on Cu (58%), COO (16%) and C=N (11%). In Schiff base ligand LUMO are distributed uniformly throughout the Me-thio group.

The frontier molecular orbitals of LH and its metal complexes obtained from B3LYP
The electronic structure of title compounds were optimized in methanol media using PCM model and then, the electronic transitions were calculated using the TD-DFT/6 31 + G(d,p) method. The experimental and theoretical electronic spectra along with the major excitation of the frontier orbitals character were given in Tables 4, 5, 6, 7, 8, and 9.
The oscillator strengths (f) and dominant excitation character for the Mn(II)-L complex. The calculated absorption spectra in the UV–Vis region are compared together with the experimental bands and the results tabulate in Tables 4, 5, 6, 7, 8, and 9. A good agreements were observed between experimental and theoretical transitions.
The calculated spectrum of the complexes associated with the frontier orbital contributions for major transitions were shown in Fig. 6. The calculated LH spectrum consist of one red shifted band at 287.4 nm (exp: 274 nm), which is assigned to π → π* transition of the aromatic thiophene group. After coordination of the ligand with Co, Ni, and Cu ions, d–d excitation bands were appeared at region of visible, while those of peaks were not at all observed for Mn(II) and Zn(II) complexes due to spin forbidden transition of d5 and d10 electronic configurations of the distorted tetrahedral complexes, respectively [41]. In the 550–900 nm region, the calculated spectrum of complex of Co(II) with d7 high spin electronic configuration showed three transitions at 780.8, 677.9, and 558.1 nm, as well as two highest energy bands at 333.9 and 277.8 nm corresponding to d–d transitions, n → π* and π → π*, respectively, whereas the spectrum of Ni(II) complex with d8 configuration is characterized by two d–d transitions in the visible region (558.2 nm and 521.7 nm) and a strong band at 334.7 nm corresponding to π → π* transition. On the other hand, the Cu(II) complex is included to a band at 703.1 nm assigned to 2T2 → 2E transition and also a high bands at 523.9 nm attributed to ligand to metal charge trasfer (LMCT) with the H-3(β) → LUMO(β) excitaion.

The calculated absorption spectra of Schiff base LH and its complexes with Mn(II), Co(II), Ni(II), Cu(II), and Zn(II)
Conclusion
In this work, the synthesis and characterization of a new bi-dentate Schiff base derived from l-methionine amino acid, and 2-acetylthiophene were investigated by experimental and theoretical approaches. It is tentatively proposed that the Schiff base LH coordinates through the nitrogen of imine moiety and the oxygen of the carboxylic group, forming a stable chelate ring structure. Based on the above interpretations, the tetrahedral structures for all complexes are proposed with general formula [ML(H2O)Cl] (where M = Mn(II), Co(II), Ni(II), Cu(II), and Zn(II)].
Furthermore, the electronic structures of the LH ligand and also Mn(II)-L, Co(II)-L, Ni(II)-L, Cu(II)-L, and Zn(II)-L complexes were optimized by DFT calculations. The obtained geometry parameters confirmed the deprotonation of the carboxylic group upon coordination with the metal ions and also longer bond lengths of imine –C=N– group in all complexes. The frequency calculations were implemented to the assignment of the fundamental vibrational modes of title compounds. The obtained results are in good agreement with the experimental data. The natural charge of the compounds together with electronic configuration and interaction energy were calculated with NBO analysis. The predicted electronic absorption spectra were achieved by TDDFT/PCM method in methanol solution suggesting the complexation of the Schiff base LH. Also the absorption bands with the major excitation characters and the frontier orbital contributions were investigated by TDDFT calculations.
References
P. Sivakumar, S. Duraisamy, G. Sundaram, K. Ramasamy, H. Takeshi, E. Akira, N. Karuppannan, Polyhedron 34, 143–148 (2012)
D. Yadav, S.N. Pandey, S. Kumar, S. Sinha, Int. J. Curr. Pharm. Rev. Res. 1, 33–46 (2011)
G.A. Balaban, İ. Özsen, H. Alyar, S. Alyar, N. Özbek, J. Mol. Struct. 1120, 259–266 (2016)
S. Chen, Y. Liu, Y. Ma, W. Qiu, Z. Sun, Adv. Func. Mater. 15, 1541–1546 (2005)
D.P. Singh, V. Malik, R. Kumar, K.K. Rasayan, J. Chem. 2, 133–138 (2009)
M.B. Oliver, A. Tasada, J.J. Fiol, A.G. Raso, A. Terron, E. Molins, Polyhedron 25, 71–80 (2006)
J. Samir, J. Therm, Anal. Calorim. 121, 1309–1319 (2015)
B. Bouzerafa, A. Ourari, D. Aggoun, R. Ruiz-Rosas, Y. Ouennoughi, E. Morallon, Res. Chem. Intermed. 42, 4839–4858 (2016)
S. Sarkar, A. Mondal, M.S. El Fallah, J. Ribas, D. Chopra, E.H. Stoeckli, Polyhedron 25, 25–30 (2006)
B. Sreenivasulu, J.J. Vittal, Inorg. Chim. Acta 362, 2735–2743 (2009)
L.A. Saghatforoush, A. Aminkhani, F. Chalabian, Trans. Met. Chem. 34, 899–904 (2009)
A.S. Patil, H.V. Naika, D.A. Kulkarnia, S.P. Badami, Spectrochim. Acta Part A 75, 347–356 (2010)
K.B. Singh, A. Prakash, K.H. Rajour, N. Bhojak, D. Adhikar, Spectrochim. Acta Part A 76, 376–383 (2010)
W.J. Geary, Coord. Chem. Rev. 7, 81–122 (1971)
A. Samir, J. Therm. Anal. Calorim. 121, 1309–1319 (2015)
Z. Chen, H. Morimoto, S. Matsunaga, M. Shibasaki, J. Am. Chem. Soc. 130, 2170–2181 (2008)
R. Janupally, B. Medepi, P.B. Devi, P. Suryadevara, V.U. Jeankumar, P. Kulkarni, P. Yogeeswari, D. Sriram, Chem. Biol. Drug Des. 86, 918–925 (2015)
J.S. Gancheff, A. Acostaa, D. Armentano, G.D. Munno, R. Chiozzonea, R. González, Inorg. Chim. Acta 387, 314–320 (2012)
O. Tamer, D. Avc, Y. Atalay, Spectrochim. Acta A 117, 78–86 (2014)
F. Weinhold, C.R. Landis, Valency and Bonding: A Natural Bond Orbital Donor-A Natural Bond Orbital Donor-Acceptor Perspective (Cambridge University Press, Cambridge, 2005)
M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G.A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H.P. Hratchian, A.F. Izmaylov, J. Bloino, G. Zheng, J.L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M.I. Shida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J.A. Montgomery Jr., J.E. Peralta, F. Ogliaro, M. Bearpark, J.J. Heyd, E. Brothers, K.N. Kudin, V.N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J.C. Burant, S.S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N.J. Millam, M. Klene, J.E. Knox, J.B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R.E. Stratmann, O. Yazyev, A.J. Austin, R. Cammi, C. Pomelli, J.W. Ochterski, R.L. Martin, K. Morokuma, V.G. Zakrzewski, G.A. Voth, P. Salvador, J.J. Dannenberg, S. Dapprich, A.D. Daniels, O. Farkas, J.B. Foresman, J.V. Ortiz, J. Cioslowski, D.J. Fox, Gaussian, Inc., Wallingford CT Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford CT, (2009)
C. Lee, W. Yang, R.G. Parr, Phys. Rev. B 37, 785–789 (1988)
P.S. Anthony, L. Radom, J. Phys. Chem. 100, 16502–16513 (1996)
K. Wolinski, J.F. Hinton, P. Pulay, J. Am. Chem. Soc. 112, 8251–8260 (1990)
J. Tomasi, B. Mennucci, R. Cammi, Chem. Rev. 105, 2999–3094 (2005)
J. Sanmartin, M.R. Bermejo, A.M. Deibe, M. Maneiro, C. Lage, A.J. Filho, Polyhedron 19, 185–192 (2000)
N.B. Arslan, N. Ozdemir, O. Dayan, N. Dege, M. Koparır, P. Koparır, H. Muglu, Chem. Phys. 439, 1–11 (2014)
T.C. Morill, Spectrometric Identification of Organic Compounds, 4thed (Wiley, New York, 1981)
K. Nakamoto, Infrared and Raman spectra of Inorganic and Coordination Compounds, 3rd edn. (Wiley, New York, 1997)
I. Grecu, R. Sandulescu, M. Neamtu, Rev. Chim. 37, 589–595 (1986)
W. Szcze, M.P. Panik, P. Stefanowicz, M.K. Kalminska, N.D. Amelis, A.O. Mjkut, A. Staszewska, M. Ratajska, M.G. Bojczuk, Polyhedron 30, 9–15 (2011)
D. Sutton, Electronic Spectra of Transition Metal Complexes, Mc (Graw-Hill, London, 1968), p. 388
A.B.P. Lever, Inorganic Electronic Spectroscopy (Elsevier Publishing Company, New York, 1968)
J.C. Bailar, H.J. Emeleus, S.R. Nyholm, A.F.T. Dickenson, Comprehensive Inorganic Chemistry (Pergamon Press, NewYork, 1975)
N.N. Greenwood, A. Earnshaw, Chemistry of the Elements (Pergamon Press, Oxford, 1984)
H. Zhang, Y. Sun, X. Chen, X. Yan, B. Sun, J. Cryst. Growth 324, 196–200 (2011)
F.A. Mulder, M. Filatov, Chem. Soc. Rev. 39, 578–590 (2010)
N. Abdel-Ghani, A.M. Mansour, M.F. Abo El-Ghar, O.M. El-Borady, H. Shorafa, Inorg. Chim. Acta 435, 187–193 (2015)
N. Hasani, A. Eslami, Polyhedron 85, 412–428 (2015)
J.C.C. Chan, P.J. Wilson, S.C.F. Au-Yeung, G.A. Webb, J. Phys. Chem. A 101, 4196–4201 (1997)
S. Sagdinc, H. Pir, Spectrochim. Acta A 73, 181–187 (2009)
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Hasani, N., Najim Abid AL-jibouri, M. Synthesis, characterization, and DFT study of some transition metal complexes with Schiff base derived from 2-acetylthiophene and l-methionine. Res Chem Intermed 43, 4585–4610 (2017). https://doi.org/10.1007/s11164-017-2898-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-017-2898-3