
Abstract
Since 1994, we have been studying an extended kindred with 105 subjects (over 8 generations) residing in Itabaianinha County, in the Brazilian state of Sergipe, who have severe isolated GH deficiency (IGHD) due to a homozygous inactivating mutation (c.57 + 1G > A) in the GH releasing hormone (GHRH) receptor (GHRHR) gene. Most of these individuals have never received GH replacement therapy. They have low GH, and very low and often undetectable levels of serum IGF-I. Their principal physical findings are proportionate short stature, doll facies, high-pitched-voice, central obesity, wrinkled skin, and youthful hair with delayed pigmentation, and virtual absence of graying. The newborns from this cohort are of normal size, indicating that GH is not needed for intra-uterine growth. However, these IGHD individuals exhibit a myriad of phenotypic changes throughout the body, with a greater number of beneficial than harmful consequences. This GHRH signal disruption syndrome has been a valuable model to study the GH roles in body size and function. This reviews summarized the findings we have reported on this cohort.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Growth hormone (GH) secretion by the pituitary is under stimulatory (GHRH, and ghrelin) and inhibitory (somatostatin and IGF-I) signals that regulate its nycthemeral production, a landmark evolutionary achievement in vertebrates. GH induces the production of the insulin like growth factor type I (IGF-I) (previously called Somatomedin C), the principal mediator of its actions, mainly in the liver, but also in several peripheral tissues. Although the somatomedin hypothesis is well established, the knowledge of the functioning of the GH/IGFs system is still incomplete. For example, the role of the insulin like growth factor type II (IGF-II) is still largely unknown. IGF-II regulates fetal development and differentiation, but its role in adults, in whom it circulates in a molar ratio of 3:1 in relation to IGF-I, is less well-understood [1]. GH is not only involved in attainment of normal body size (bone and soft tissues growth), but also in the regulation of key body functions, including, among others, metabolism and cardiovascular function; musculoskeletal function and balance; sensory perception; immune function; sleep, quality of life and reproduction; DNA repair and cancer risk. These key functions relate to general health or morbidity and ultimately to longevity [2].
Studying GH deficiency (GHD) can be used to asses GH functions, but this approach has its caveats. Idiopathic GHD, important cause of short stature in childhood, can disappear in adulthood, posing questions about its nature or relevance. On other hand, acquired GHD is often caused by hypopituitarism of different etiologies, mostly pituitary tumors and surgery or irradiation, often associated with other pituitary hormones deficit, with lack or inadequacy of respective replacement therapies. Genetic isolated GHD (IGHD) can be an alternative way to assess the biological impacts of GH, but it is difficult to have a number of affected individuals, particularly naïve to GH treatment.
Since 1994 we have been studying, an extended kindred with 105 IGHD subjects (alive or dead) over eight generations, residing in Itabaianinha county, in the northeastern Brazilian state of Sergipe. All the affected individuals are homozygous for the same null c.57 + 1G > A inactivating mutation in the GH releasing hormone (GHRH) receptor (GHRHR) gene (GHRHR. OMIM n. 612,781), with a founder effect [3]. These subjects previously lived in the village of Carretéis and its rural outskirts, an area surrounded by mountains. The geographical isolation, the high frequency of consanguineous unions, their historical zero mobility and normal longevity were the principal causes for the spreading of this mutation in this particular area [4,5,6]. Presently, most of the IGHD individuals have moved to the municipality of Itabaianinha, while eleven of them moved to other cities. The increased mobility of the affected IGHD subjects and of the heterozygous and the increasing awareness of its genetic cause attenuate the chance of the meeting of mutated alleles, reducing the birth of the homozygous newborns. This, and the current, albeit inconsistent, treatment of IGHD children makes the thorough study of this cohort even more important, as it will not last forever. Presently we are aware of 54 living IGHD individuals in this cohort.
This IGHD cohort presents low but detectable GH secretion, with values of serum GH peak lower than 1 ng/ml in both clonidine and insulin tolerance tests, and no response to GHRH [7]. This is combined with life-long severe reduction of circulating IGF-I, and considerable IGF-II up-regulation, proven by an increase in the IGF-II/IGFBP-3 ratio, a measure of its bioavailability [2, 5, 8]. We hypothesize that this IGF-II up-regulation may have physiological implications, contributing to the IGF bioavailability to some vital tissues like the brain and the eye, in which growth seems to be less dependent from pituitary GH and circulating IGF-I [5]. These subjects also have higher serum cortisol concentrations (likely due to the increased activity of the enzyme 11 beta-hydroxysteroid dehydrogenase, which converts cortisone to cortisol) [9]. Other hormonal findings includes a moderate increase in serum TSH levels (likely due to a reduced effect of IGF-I on hypothalamic somatostatin) [10], a reduction in serum total T3 and an increase of serum free T4 (likely due to a reduction in the function of the GH-dependent deiodinase system) [11]. A mild increase of total testosterone (due to an increase in SHBG) was also observed in males [12]. Table 1 summarizes the hormonal findings of this GHRH disruption syndrome.
The principal physical findings of these adult subjects are proportionate short stature, doll facies, high-pitched-voice, central obesity, wrinkled skin, and youthful hair with delayed pigmentation in children and teenagers, and virtual absence of graying, even in old age (Table 2). However, these IGHD individuals exhibit a myriad of phenotypic variants throughout the body, with a greater number of beneficial than harmful consequences to their overall health, and with positive consequences on quality of life [13], and possibly longevity [6]. In this review, we describe this particular disruption of the GHRH signaling and its impact in children and adults and its consequences on body size (Table 3) and function (Table 4).
1 Body size in IGHD
1.1 Bone growth
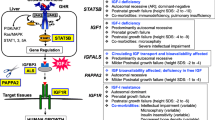
It is intuitive that an adequate body size and composition provides an evolutionary advantage in terms of obtaining food and reproducing. However, normal growth involves some costs in terms of aging and longevity [2]. Therefore, a small body size can make these two aspects compatible. We will analyze this assumption in our IGHD model due to GHRH disruption (Fig. 1).

Two married IGHD individuals, homozygous for the mutation.57 + 1G > A GHRHR mutation. The man is the tallest IGHD individual (56 years and 137 cm), together with his wife (58 years and 121 cm). The control with normal height is an author, Dr. M.H. A-O (61 years and 174 cm). The main physical findings of these IGHD individuals are proportionate short stature, doll facies, high-pitched voice, central obesity and wrinkled skin
Differently from newborns with Laron syndrome (due to GH resistance caused by mutations in the GH receptor), who have mildly reduced birth length and birth weight [14], newborns from this IGHD cohort are of normal size, suggesting that the GH resistance has a greater impact on birth size than GHRH resistance. In the Itabaianinha cohort postnatal growth failure becomes evident at about 1.5 years of age, with height standard deviation scores (SDS) of −3.2 and it is progressive, with adult stature SDS range from −5.1 to −9.6 [12]. Adult height in untreated individuals is 128.7 ± 5.9 cm in males (range 117–137 cm), and 117.6 ± 5.7 cm in females (range 107–126 cm) [15]. We speculate that birth size in our IGHD model is kept normal by IGF-II, insulin, or placental factors (placental GH, and or placental lactogens) [16], as previously suggested in men [17] or in mice [18]. These data indicate that GH is not truly a perennial growth hormone, but a potent post-natal elongation factor.
Curiously, the consequences of IGHD on bone growth are uneven, with a gradient of reduction of the mean SDS: stature (−7.0), maxillary length (−6.5), anterior facial height (−4.3), and head circumference (−2.7) [5, 19, 20]. The greater impact of IGHD on adult height than on head size may reflect the fact that much of the growth of the brain and the skull occurs early in life, when growth is less dependent on pituitary GH, while the elongation of the rest of the body is mainly postnatal and strongly dependent on GH and endocrine IGF-I. Relative increased brain size is also found in GH resistance syndromes both in men [21] and mice [2]. The disproportion between the reduction of calvarium and the facial height results in one typical feature of the IGHD syndrome, the doll-like or “cherubim angel”facies. The cephalometric features together with the underdevelopment of the larynx lead to another paramount physical finding of congenital IGHD, the high-pitched voice with high fundamental frequency in both genders, abolishing the effects of puberty and aging on the voice [20, 22, 23]. These data indicate that the consequences of IGHD on bone development are not uniform, and probably influenced by local factors, according to a physiological hierarchy [24]. For instance, brain and skull sizes seem more relevant than body size. Consequently, the growth of the brain may be more preserved than somatic growth.
1.2 Soft tissues growth
The growth of soft tissues is also unevenly affected, suggesting tissue-specific consequences of IGHD. When corrected by body surface area (BSA), the thyroid, spleen and uterus volumes [11], and the left ventricular mass [25] are significantly smaller than those of normal controls, indicating a greater dependency on the pituitary GH or circulating IGF-I. On the other hand, BSA-corrected prostate and ovaries volumes are normal, while pancreas, liver, and kidneys appear larger compared to controls [26], suggesting a reduced dependence from GH for the growth of these three organs. Probably these organs have local compensator mechanisms. This discrepancy in relative sizes includes several structures or organs. We will highlight three, the pituitary, the eye and the pharynx.
IGHD subjects with GHRHR mutations exhibit marked anterior pituitary hypoplasia [27], due to the lack of the GHRH effect on the expansion of somatotroph cells, without abnormalities of the stalk or neuro-hypophysis, similar to the findings in mice with a mutation in the same gene (little mouse) [28]. On other hand, the non-corrected ocular axial length corresponds to 96% of the normal controls, similar to the head circumference (93%), while stature corresponds to 75%, suggesting a parallelism between eye and head growth [29]. Adequate eye and head (brain) development likely represent important elements of the environmental adaptation and survival capacity of these individuals. Eye and brain growth may involve different patterns of temporal regulation than the whole body growth, reflecting a physiologic hierarchy controlling body size and function and showing a lesser dependency on pituitary GH or endocrine IGF-I for their growth [5]. Accordingly, the brain completes 83.6% of its growth within the first year of life, with essentially full growth achieved during the first three years of life [30]. Very recently, we have shown that the pharyngeal airway width in IGHD adults is similar to normal controls, showing that this structure is less dependent from the pituitary GH and circulating IGF-I [31]. This finding contributes to the low prevalence of obstructive sleep apnea syndrome in these subjects [32].
The external genitalia of these IGHD subjects is essentially normal sized. Accordingly, most of the marriages of the IGHD subjects, independently of gender, occur with normal statured partners [5]. In conclusion, the growth of different tissues growth, cannot be fully explained by the pituitary GH/circulating IGF-I axis.
2 Body function in IGHD
2.1 Metabolism and cardiovascular function
As mentioned earlier, GH is a potent post-natal elongation factor in humans. To accomplish this task, GH has important roles in the processes of getting food and thereby energy, utilizing or storing it, by a complex system of enteroendocrine connections.
This network includes GH, a hyperglycemic, anabolic and lipolytic peptide, and IGF-I, a hypoglycemic, anabolic, and lipotropic agent. In addition, GHRH, ghrelin, somatostatin, insulin and gastrointestinal hormones including GLP-1 are involved in this network. Ghrelin, an orexigenic peptide and potent stimulatory GH factor, is produced mostly by the stomach, and enhances feeding and weight gain. Insulin and GH are essential in homoeothermic animals to adapt the energy utilization to the amount of food, promoting anabolism when calorie supply exceeds demands, and catabolism in the opposite situation. Insulin is the main metabolic hormone in the fed state, but GH assumes a key role as a stimulator of lipolysis during prolonged fasting, providing an evolutionary advantage in times of food scarcity [33]. Further, the ghrelin-GH axis seems have an essential role for surviving starvation, at least in mice [34]. IGHD subjects with this GHRHR mutation eat proportionally more, but healthier than local controls, matched for age and gender. In fact, their estimated energy intake corrected by body weight was higher than controls. In addition, they consume in percentage more proteins less carbohydrates, and equal amount of lipids in comparison to controls [35]. More recently, we demonstrated that this IGHD syndrome is associated with increased GLP-1 secretion and reduced postprandial ghrelin and hunger attenuation in response to a mixed meal [36]. These enteroendocrine connections can result into a favorable outcome in terms of environmental adaptation, guaranteeing appropriate food intake, and can confer metabolic and vascular benefits [36].
These IGHD subjects have higher locomotor activity [37] and possible increased thermogenesis, similar to what was shown in mice with generalized ablation of the GHRH gene [38], factors that can contribute to their partial protection against weight gain. Because they have a reduced sweating capacity, they may have a disadvantage in the tropical climate of their region [39].
The changes in body composition in our IGHD cohort are already present in early childhood, and persist throughout life. They include decreased fat free mass and increased percent body fat [40, 41]. Obesity is predominantly truncal, likely due to reduced lipolysis by the GH-sensitive lipase in this body region [9]. Truncal obesity may be a direct effect of the lack of GH, as IGF-I treatment does not reverse it in subjects with GH insensitivity (Laron) syndrome (GHI) [42]. Obesity seems to be less pronounced in IGHD than in GHI syndrome [39], probably reflecting some residual lipolysis in IGHD due to the low, but detectable, GH secretion. In spite of visceral adiposity, IGHD subjects from our cohort have increased insulin sensitivity [43], accompanied by high serum adiponectin [44]. The increased insulin sensitivity despite visceral adiposity suggests that there is a threshold of GH secretion necessary for visceral adiposity to impair insulin sensitivity [42]. Similar insulin sensitivity and increased adiponectin is seen in GH resistant mice [45, 46]. The insulin sensitivity may contribute to the normal longevity despite increased cholesterol of our IGHD cohort [6]. However, the insulin sensitivity does not prevent the development of diabetes, likely due to reduced β-cell function [45].
These IGHD subjects have, throughout life, high serum total and LDL cholesterol levels [25, 47], secondary to a decrease in hepatic LDL receptor activity which is caused by GHD [48]. They also have higher circulating C-reactive protein, with a mild increase in systolic blood pressure in adults, and arterial hypertension in older age, without evidence of cardiac hypertrophy or increase in carotid intima media thickness, or coronary [49, 50] and abdominal aortic atherosclerosis [51]. This observation may be explained by the fact that IGF-I has a dual action, promoting (by increasing proliferation of vascular smooth muscle cells), or preventing (by increasing nitric oxide formation, vascular compliance, and insulin sensitivity) atherogenesis. A persistent very low IGF-I level might have a protective role, whereas a milder decrease (as seen in adult-onset GH-deficiency) might be noxious [49]. Indeed, when we partially reversed their GHD with a 6-month GH treatment, we observed the appearance of carotid plaques [52].
Nonalcoholic fatty liver disease (NAFLD) is more prevalent in the IGHD adults than local controls, without progress to advanced forms of hepatitis, challenging the concept of a direct role of GH/IGF-I deficiency in the pathogenesis of advanced NAFLD stages in acquired hypopituitarism [53].
2.2 Musculoskeletal function and balance
Understandably, individuals with IGHD have small bones and muscles. However, they are so well adapted (mechanosthatic theory) that their volumetric bone mineral density, corrected by the size of the bone, is similar to controls [54]. In addition, they have better muscle strength parameters (adjusted for weight and fat free mass), and greater peripheral resistance to fatigue than controls [37]. They have normal levels of 25 hydroxyvitamin D and normal phosphor-calcium homeostasis [39]. Not surprisingly, there has not been any report of spontaneous fractures in this cohort, and the prevalence of vertebral fracture is indeed reduced in older IGHD individuals compared to age-matched controls [51]. These subjects exhibit satisfactory walking and postural balance, without increase in fall risk [55]. More recently, we reported that they have higher prevalence of moderate peripheral vestibular impairment, and of abnormal vestibular-ocular reflex, without relevant clinical consequences, allowing them to be quite active in farming, horse riding, and sports [16].
2.3 Sensory perception
The sensory perceptions are extremely important for environmental adaptation. Compared to normal controls, these IGHD subjects exhibit similar visual acuity, intraocular pressure and lens thickness, but higher values of spherical equivalent and corneal curvature and lower measures of axial length, anterior chamber depth, and central corneal thickness [29]. They also have an increase of optic disc size, but with similar thickness of the macula, and moderate reduction of vascular retinal branching points [56]. Taken together, eye changes translate in little, if any, vision impairment. Conversely, the inner ear is affected by GHD, with changes in cochlea (mild high-tone sensorineural hearing loss) [57] and labyrinth (moderate peripheral vestibular impairment) [16].
2.4 Immune function
The immune function is also very important for environmental adaptation and survival capacity. Mice with ablation of the GHRH gene present spleen atrophy and a severe deficiency in vaccine and immune responses against Streptococcus pneumonia [58]. Although we have not yet studied the response of these IGHD subjects to the immunization against this specific pathogen, we have not observed significant immune deficits in this cohort. We have studied the frequency of infectious diseases and the immune function via a clinical questionnaire, physical exam, serology, and serum total IgG, IgM, IgE and IgA measurement. The immune response was evaluated by skin tests and response to vaccination for hepatitis B, tetanus, and bacillus Calmette-Guérin. There was no difference between IGHD and controls in history of infectious diseases and baseline serology. IGHD subjects had lower total IgG than controls, but within normal range. There was no difference in the response to any of the vaccinations, or in the positivity to PPD, streptokinase or candidin [59]. On other hand, they have higher prevalence of periodontal disease than local controls [60]. The apparently normal daily immune function suggests that many immune cells have a local GH/IGF system, apparently independent from the pituitary GH and endocrine IGF-I. Very recently, we found that macrophages from IGHD subjects are less prone to Leishmania infection compared to GH sufficient controls [61]. These findings may indicate a genetic advantage of this IGHD cohort against at least this particular pathogen, which is prevalent in northeast Brazil, and may have contributed to the spreading of the mutation in this area.
2.5 Sleep, quality of life and reproduction
GHRH and GH play an important role in sleep regulation, and GHD has been associated with sleep abnormalities [62]. Indeed, our IGHD individuals exhibit impairment in both non-REM and REM sleep, more conspicuous in the former [32], but this does not affect their quality of life (QoL) measured by a standardized, self-weighted questionnaire [13]. This contrast with many reports of reduced QoL in adults with acquired GHD. We hypothesize that having very little GH exposure throughout life may be more advantageous than having normal GH secretion followed by subsequent decline.
Subjects with IGHD have no obvious abnormalities in sexual development or function. They do not exhibit microphallus, but have delayed puberty. The beginning of climacteric is anticipated, but the age at menopause is similar to control group, and within the age range of normality. The IGHD women have a smaller number of children than other women from the same area [5, 63], but this may be related to becoming sexually active at a later age, and to the need for caesarian deliveries due to cephalic/pelvic disproportion. They do not have trouble in breast-feeding. Although GH and prolactin share strong similarities in structure and function, adequate mammary development and lactation are apparently possible in the absence of GH signals [12, 63].
2.6 DNA repair and Cancer
In the whole IGHD Itabaianinha cohort, during 25 years of medical care, our team did not diagnose any case of breast, colon, and prostate cancers [2, 5]. The absence of these common neoplasms suggests that GH and IGF-I deficiency protects against DNA damage and favors apoptosis of damaged cells, thereby reducing the risk of cancer, as shown in Laron patients [64, 65]. However, the deficiency of GH/IGF-I may anticipate errors in the repair of potentially mutagenic DNA damage in human skin keratinocytes. Accordingly, there were four skin tumors; three of them were malignant (one lethal), indicating a vulnerability of their skin to tumor development [66].
2.7 Longevity
In many animal models GH deficiency or resistance are associated with increased lifespan (2). Although the IGHD subjects in our cohort may have deleterious age-related conditions, such as diabetes, NAFLD, and skin cancer, these conditions very rarely progress to advanced stages, compromising their normal longevity, with some centenarians reported among the IGHD subjects [2, 6]. While the increased insulin sensitivity is one plausible explanation for this beneficial profile, studies of other putative mechanisms, such as mTOR pathway or mitochondrial function in these IGHD subjects are necessary. However, our data indicate that these subjects present less tiredness and disabilities even in advanced age, and consequently old subjects exhibit a different profile of senescence, maintaining a more youthful level of morbidity [2]. Not surprisingly, evitable causes of mortality, such as accidents, seem more prevalent in the IGHD cohort than in general Itabaianinha population, possibly due to disproportion between these subjects’ size and commonly used tools that are designed for normal statured individuals [2].
3 Conclusions
This GHRH disruption syndrome in the Itabaianinha cohort has been a very valuable model to study the GH/IGF-I axis roles in body size and function. Although much progress has been achieved, this knowledge is still incomplete. However, these very small-sized individuals seem to have more positive than harmful consequences in their body functions, with a beneficial result in terms of quality of life, without reducing their longevity.
References
Livingstone C, Borai A. Insulin-like growth factor-II: its role in metabolic and endocrine disease. Clin Endocrinol. 2014;80:773–81.
Aguiar-Oliveira MH, Bartke A. Growth hormone deficiency: health and longevity. Endocr Rev. 2019;40:575–601.
Salvatori R, Hayashida CY, Aguiar-Oliveira MH, Phillips JA III, Souza AH, Gondo RG, et al. Familial isolated growth hormone deficiency due to a novel mutation in the growth hormone releasing hormone receptor. J Clin Endocrinol. 1999;84:917–23.
Souza AH, Salvatori R, Martinelli CE Jr, Carvalho WM, Menezes CA, Barretto ES, et al. Growth or somatotrophic hormone: new perspectives in isolated GH deficiency after description of the mutation in the GHRH receptor gene in individuals of Itabaianinha County, Brazil. Arq Bras Endocrinol Metabol. 2004;48:406–13.
Aguiar-Oliveira MH, Souza AHO, Oliveira CRP, Campos VC, Oliveira-Neto LA, Salvatori R. MECHANISMS IN ENDOCRINOLOGY: the multiple facets of GHRH/GH/IGF-I axis: lessons from lifetime, untreated, isolated GH deficiency due to a GHRH receptor gene mutation. Eur J Endocrinol. 2017;177:R85–97.
Aguiar-Oliveira MH, Oliveira FT, Pereira RM, Oliveira CR, Blackford A, Valenca EH, et al. Longevity in untreated congenital growth hormone deficiency due to a homozygous mutation in the GHRH receptor gene. J Clin Endocrinol Metab. 2010;95:714–21.
Salvatori R, Serpa MG, Parmigiani G, Britto AV, Oliveira JL, Oliveira CR, et al. GH response to hypoglycemia and clonidine in the GH-releasing hormone resistance syndrome. J Endocrinol Investig. 2006;29:805–8.
Aguiar-Oliveira MH, Gill MS, de Barretto AES, Alcântara MR, Miraki-Moud F, Menezes CA, et al. Effect of severe growth hormone (GH) deficiency due to a mutation in the GH-releasing hormone receptor on insulin-like growth factors (IGFs), IGF-binding proteins, and ternary complex formation throughout life. J Clin Endocrinol Metab. 1999;84:4118–26.
Gomes-Santos E, Salvatori R, Ferrao TO, Oliveira CR, Diniz RD, Santana JA, et al. Increased visceral adiposity and cortisol to cortisone ratio in adults with congenital lifetime isolated GH deficiency. J Clin Endocrinol Metab. 2014;99:3285–9.
Gondo RG, Aguiar-Oliveira MH, Hayashida CY, Toledo SP, Abelin N, Levine MA, et al. Growth hormone-releasing peptide-2 stimulates GH secretion in GH-deficient patients with mutated GH-releasing hormone receptor. J Clin Endocrinol Metab. 2001;86:3279–83.
Alcântara MR, Salvatori R, Alcântara PR, Nóbrega LM, Campos VS, Oliveira EC, et al. Thyroid morphology and function in adults with untreated isolated growth hormone deficiency. J Clin Endocrinol Metab. 2006;91:860–4.
Menezes M, Salvatori R, Melo LD, Rocha IE, Oliveira CR, Pereira RM, et al. Prolactin and sex steroids levels in congenital lifetime isolated GH deficiency. Endocrine. 2013;44:207–11.
Barbosa JA, Salvatori R, Oliveira CR, Pereira RM, Farias CT, Britto AV, et al. Quality of life in congenital, untreated, lifetime isolated growth hormone deficiency. Psychoneuroendocrinology. 2009;34:894–900.
Savage MO, Blum WF, Ranke MB, Postel-Vinay MC, Cotterill AM, Hall K, et al. Clinical features and endocrine status in patients with growth hormone insensitivity (Laron syndrome). J Clin Endocrinol Metab. 1993;77:1465–71.
Aguiar-Oliveira MH, Salvatori R. Lifetime growth hormone deficiency: Impact on growth, metabolism, body composition, and survival capacity. In: Preedy VR, ed. Handbook of growth and growth monitoring in health and disease New York: Springer; 2012:2699–2710.
Santos-Carvalho HA, Aguiar-Oliveira MH, Salvatori R, Valença EHO, Andrade-Guimarães AL, Palanch-Repeke CE, et al. Vestibular function in severe GH deficiency due to an inactivating mutation in the GH releasing hormone receptor gene. Endocrine. 2020. https://doi.org/10.1007/s12020-019-02178-3.
Handwerger S, Freemark M. The roles of placental growth hormone and placental lactogens in the regulation of human fetal growth and development. J Pediatr Endocrinol Metab. 2000;13:343–56.
Liu J, Barker J, Perkins A, Robertson E, Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf.1) and type 1 IGF receptor (Igflr). Cell. 1993;75:59–72.
Oliveira-Neto LA, Melo Mde F, Franco AA, Oliveira AH, Souza AH, Valenca EH, et al. Cephalometric features in isolated growth hormone deficiency. Angle Orthod. 2011;81:578–83.
Valenca EH, Salvatori R, Souza AH, Oliveira-Neto LA, Oliveira AH, Goncalves MI, et al. Voice formants in individuals with congenital, isolated, lifetime growth hormone deficiency. J Voice. 2016;30:281–6.
Laron Z, Iluz M, Kauli R. Head circumference in untreated and IGF-I treated patients with Laron syndrome: comparison with untreated and hGH-treated children with isolated growth hormone deficiency. Growth Hormon IGF Res. 2012;22(2):49–52.
Barreto VM, D'Avila JS, Sales NJ, Goncalves MI, Seabra JD, Salvatori R, et al. Laryngeal and vocal evaluation in untreated growth hormone deficient adults. Otolaryngol Head Neck Surg. 2009;140(1):37–42.
Valenca EH, Souza AH, Oliveira AH, Valenca SL, Salvatori R, Goncalves MI, et al. Voice quality in short stature with and without GH deficiency. J Voice. 2012;26:673 e613–679.
Aguiar-Oliveira MH, Davalos C, Campos VC, Oliveira Neto LA, Marinho CG, Oliveira CRP. Hypothalamic abnormalities: growth failure due to defects of the GHRH receptor. Growth Hormon IGF Res. 2018;38:14–8.
Barreto-Filho JA, Alcantara MR, Salvatori R, Barreto MA, Sousa AC, Bastos V, et al. Familial isolated growth hormone deficiency is associated with increased systolic blood pressure, central obesity, and dyslipidemia. J Clin Endocrinol Metab. 2002;87:2018–23.
Oliveira CR, Salvatori R, Nobrega LM, Carvalho EO, Menezes M, Farias CT, et al. Sizes of abdominal organs in adults with severe short stature due to severe, untreated, congenital GH deficiency caused by a homozygous mutation in the GHRH receptor gene. Clin Endocrinol (Oxf). 2008;69:153–8.
Oliveira HA, Salvatori R, Krauss MP, Oliveira CR, Silva PR, Aguiar-Oliveira MH. Magnetic resonance imaging study of pituitary morphology in subjects homozygous and heterozygous for a null mutation of the GHRH receptor gene. Eur J Endocrinol. 2003;148:427–32.
Wilson DB, Wyatt DP. Histopathology of the pituitary gland in neonatal little (lit) mutant mice. HistolHistopathol. 1992;7:451–5.
Faro ACN, Pereira-Gurgel VM, Salvatori R, Campos VC, Melo GB, Oliveira FT, et al. Ocular findings in adult subjects with an inactivating mutation in GH releasing hormone receptor gene. Growth Hormon IGF Res. 2017;34:8–12.
Nellhaus G. Head circumference from birth to eighteen years. Practical composite international and interracial graphs. Pediatrics. 1968;41:106–14.
Reinheimer DM, Andrade BMR, Nascimento JKF, Fonte JBM, Araújo IMP, Martins-Filho PRS, et al. Formant frequencies, cephalometric measures, and pharyngeal airway width in adults with congenital, isolated, and untreated growth hormone deficiency. J Voice. 2019;27. https://doi.org/10.1016/j.jvoice.2019.04.014.
Oliveira FT, Salvatori R, Marcondes J, Macena LB, Oliveira-Santos AA, Faro ACN, et al. Altered sleep patterns in patients with non-functional GHRH receptor. Eur J Endocrinol. 2017;177:51–7.
Sakharova AA, Horowitz JF, Surya S, Goldenberg N, Harber MP, Symons K, et al. Role of growth hormone in regulating lipolysis, proteolysis, and hepatic glucose production during fasting. J Clin Endocrinol Metab. 2008;93:2755–9.
Goldstein JL, Zhao TJ, Li RL, Sherbet DP, Liang G, Brown MS. Surviving starvation: essential role of the ghrelin-growth hormone axis. Cold Spring HarbSymp Quant Biol. 2011;76:121–7.
Oliveira-Santos AA, Salvatori R, Gomes-Santos E, Santana JA, Leal AC, Barbosa RA, et al. Subjects with isolated GH deficiency due to a null GHRHR mutation eat proportionally more, but healthier than controls. Endocrine. 2016;51:317–22.
Oliveira-Santos AA, Salvatori R, Nogueira MC, Bueno AC, Barros-Oliveira CS, Leal ÂCGB, et al. Enteroendocrine Connections in Congenital Isolated GH Deficiency Due to a GHRH Receptor Gene Mutation. J Clin Endocrinol Metab. 2019;104:2777–84.
Andrade-Guimarães AL, Aguiar-Oliveira MH, Salvatori R, Carvalho VO, Alvim-Pereira DCRA, GAM B, et al. Adult individuals with congenital, untreated, severe isolated growth hormone deficiency have satisfactory muscular function. Endocrine. 2019;63:112–9.
Leone S, Chiavaroli A, Shohreh R, Ferrante C, Ricciuti A, Manippa F, et al. Increased locomotor and thermogenic activity in mice with targeted ablation of GHRH geneGrowthHorm. IGF Res. 2015;25:80–4.
Barros-Oliveira CS, Salvatori R, Dos Santos JSS, Santos PFC, Oliveira-Santos AA, Marinho CG, et al. Sweat and vitamin D status in congenital, lifetime, untreated GH deficiency. Endocrine. 2019;65:710–3.
de ABES, Gill MS, De Freitas ME, Magalhaes MM, Souza AH, Aguiar-Oliveira MH, Clayton PE. Serum leptin and body composition in children with familial GH deficiency (GHD) due to a mutation in the growth hormone-releasing hormone (GHRH) receptor. ClinEndocrinol (Oxf). 1999;51:559–564.
Laron Z, Ginsberg S, Lilos P, Arbiv M, Vaisman N. Body composition in untreated adult patients with Laron syndrome (primary GH insensitivity). Clin Endocrinol. 2006;65:114–7.
Oliveira CR, Salvatori R, Barreto-Filho JA, Rocha IE, Mari A, Pereira RM, et al. Insulin sensitivity and beta-cell function in adults with lifetime, untreated isolated growth hormone deficiency. J Clin Endocrinol Metab. 2012;97:1013–9.
Oliveira CR, Salvatori R, Meneguz-Moreno RA, Aguiar-Oliveira MH, Pereira RM, Valenca EH, et al. Adipokine profile and urinary albumin excretion in isolated growth hormone deficiency. J Clin Endocrinol Metab. 2010;95:693–8.
Masternak MM, Bartke A, Wang F, Spong A, Gesing A, Fang Y, et al. Metabolic effects of intra-abdominal fat in GHRKO mice. Aging Cell. 2012;11:73–81.
Bennis MT, Schneider A, Victoria B, Do A, Wiesenborn DS, Spinel L, et al. The role of transplanted visceral fat from the long-lived growth hormone receptor knockout mice on insulin signaling. Geroscience. 2017;39:51–9.
Vicente TA, Rocha IE, Salvatori R, Oliveira CR, Pereira RM, Souza AH, et al. Lifetime congenital isolated GH deficiency does not protect from the development of diabetes. Endocr Connect. 2013;2:112–7.
Gleeson HK, Souza AH, Gill MS, Wieringa GE, Barretto ES, Barretto-Filho JA, et al. Lipid profiles in untreated severe congenital isolated growth hormone deficiency through the lifespan. Clin Endocrinol. 2002;57:89–95.
Rudling M, Olivecrona H, Eggertsen G, Angelin B. Regulation of rat hepatic low density lipoprotein receptors. In vivo stimulation by growth hormone is not mediated by insulin-like growth factor I. J Clin Invest. 1996;97:292–9.
Menezes Oliveira JL, Marques-Santos C, Barreto-Filho JA, Ximenes Filho R, de Oliveira Britto AV, Oliveira Souza AH, et al. Lack of evidence of premature atherosclerosis in untreated severe isolated growth hormone (GH) deficiency due to a GH-releasing hormone receptor mutation. J Clin Endocrinol Metab. 2006;91:2093–9.
Costa UM, Oliveira CR, Salvatori R, Barreto-Filho JA, Campos VC, Oliveira FT, et al. Brazilian adult individuals with untreated isolated GH deficiency do not have accelerated subclinical atherosclerosis. Endocr Connect. 2016;5:41–6.
Souza AH, Farias MI, Salvatori R, Silva GM, Santana JA, Pereira FA, et al. Lifetime, untreated isolated GH deficiency due to a GH-releasing hormone receptor mutation has beneficial consequences on bone status in older individuals, and does not influence their abdominal aorta calcification. Endocrine. 2014;47:191–7.
Oliveira JL, Aguiar-Oliveira MH, D'Oliveira A Jr, Pereira RM, Oliveira CR, Farias CT, et al. Congenital growth hormone (GH) deficiency and atherosclerosis: effects of GH replacement in GH-naive adults. J Clin Endocrinol Metab. 2007;92:4664–70.
Diniz RD, Souza RM, Salvatori R, Franca A, Gomes-Santos E, Ferrao TO, et al. Liver status in congenital, untreated, isolated GH deficiency. Endocr Connect. 2014;3:132–7.
Epitacio-Pereira CC, Silva GM, Salvatori R, Santana JA, Pereira FA, Gois-Junior MB, et al. Isolated GH deficiency due to a GHRH receptor mutation causes hip joint problems and genu valgum, and reduces size but not density of trabecular and mixed bone. J Clin Endocrinol Metab. 2013;98:E1710–5.
Santana-Ribeiro AA, Moreira-Brasileiro GA, Aguiar-Oliveira MH, Salvatori R, Carvalho V, Alvim-Pereira CK, et al. Walking and postural balance in adults with severe short stature due to isolated GH deficiency. Endocr Connect. 2019. https://doi.org/10.1530/EC-19-0103.
Pereira-Gurgel VM, Faro AC, Salvatori R, Chagas TA, Carvalho-Junior JF, Oliveira CR, et al. Abnormal vascular and neural retinal morphology in congenital lifetime isolated growth hormone deficiency. Growth Hormon IGF Res. 2016;30–31:11–5.
Prado-Barreto VM, Salvatori R, Santos Junior RC, Brandao-Martins MB, Correa EA, Garcez FB, et al. Hearing status in adult individuals with lifetime, untreated isolated growth hormone deficiency. Otolaryngol Head Neck Surg. 2014;150:464–71.
Farhat K, Bodart G, Charlet-Renard C, Desmet CJ, Moutschen M, Beguin Y, et al. Growth hormone (GH) deficient mice with GHRH gene ablation are severely deficient in vaccine and immune responses against Streptococcus pneumoniae. Front Immunol. 2018;2:2175. https://doi.org/10.3389/fimmu.2018.02175.
Campos VC, Barrios MR, Salvatori R, de Almeida RP, de Melo EV, Nascimento AC, et al. Infectious diseases and immunological responses in adult subjects with lifetime untreated, congenital GH deficiency. Endocrine. 2016;54:182–90.
Britto IM, Aguiar-Oliveira MH, Oliveira-Neto LA, Salvatori R, Souza AH, Araujo VP, et al. Periodontal disease in adults with untreated congenital growth hormone deficiency: a case-control study. J Clin Periodontol. 2011;38:525–31.
Barrios MR, Campos VC, Peres NTA, de Oliveira LL, Cazzaniga RA, Santos MB, et al. Macrophages From Subjects With Isolated GH/IGF-I Deficiency Due to a GHRH Receptor Gene Mutation Are Less Prone to Infection by Leishmaniaamazonensis. Front Cell Infect Microbiol. 2019;9:311. https://doi.org/10.3389/fcimb.2019.00311 eCollection 2019.
Copinschi G, Nedeltcheva A, Leproult R, Morselli LL, Spiegel K, Martino E, et al. Sleep disturbances, daytime sleepiness, and quality of life in adults with growth hormone deficiency. J Clin Endocrinol Metab. 2010;95:2195–202.
Menezes M, Salvatori R, Oliveira CR, Pereira RM, Souza AH, Nobrega LM, et al. Climacteric in untreated isolated growth hormone deficiency. Menopause. 2008;15:743–7.
Guevara-Aguirre J, Balasubramanian P, Guevara-Aguirre M, Wei M, Madia F, Cheng CW, et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci Transl Med. 2011;3:70ra13.
Steuerman R, Shevah O, Laron Z. Congenital IGF1 deficiency tends to confer protection against post-natal development of malignancies. Eur J Endocrinol. 2011;164:485–9.
Marinho CG, Mermejo LM, Salvatori R, Assirati JAJ, Oliveira CRP, Santos EG, et al. Occurrence of neoplasms in individuals with congenital, severe GH deficiency from the Itabaianinha kindred. Growth Hormon IGF Res. 2018;41:71–4.
Acknowledgements
The authors thank the IGHD individuals for the participation in all these studies and all the many collaborators (particularly Prof. Anita H. O. Souza, who performed the fundamental population genetic study), who have contributed to these studies.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
M. H. Aguiar-Oliveira has nothing to disclose. R. Salvatori serves on Novordisk advisory board.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. All the participants gave informed consent.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Aguiar-Oliveira, M.H., Salvatori, R. Disruption of the GHRH receptor and its impact on children and adults: The Itabaianinha syndrome. Rev Endocr Metab Disord 22, 81–89 (2021). https://doi.org/10.1007/s11154-020-09591-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11154-020-09591-4