
Abstract
Glucokinase is a key enzyme involved in regulating insulin secretion from the pancreatic ß-cell. The unique role of glucokinase in human glucose physiology is illustrated by the fact that genetic mutations in glucokinase can either cause hyperglycaemia or hypoglycaemia. Heterozygous inactivating mutations in glucokinase cause maturity-onset diabetes of the young (MODY), homozygous inactivating in glucokinase mutations result in permanent neonatal diabetes whereas heterozygous activating glucokinase mutations cause hyperinsulinaemic hypoglycaemia
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The hexokinase group of enzymes (of which there are 4, designated hexokinases I, II, III, and IV or hexokinases A, B, C, and D) phosphorylate a six carbon sugar (such as hexose) to hexose phosphate. Glucokinase (hexokinase IV or D) is a member of the hexokinase family of enzymes but it is distinct (in terms of enzyme kinetics) from the other three groups [1]. The name, glucokinase, is derived from its relative specificity for glucose under physiologic conditions. The expression of glucokinase is limited to the major organs (such as the pancreas, liver, brain and the gastrointestinal tract) which are thought to have an integrated role in glucose sensing [2].
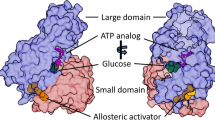
Glucokinase is a key regulatory enzyme in the pancreatic ß-cells [3]. It operates as a monomer and phosphorylates glucose on carbon 6 with MgATP as a second substrate to form glucose-6-phosphate (G6P) as a first step in the glycolytic pathway (Fig. 1). In the liver the phosphorylation of glucose by glucokinase promotes glycogen synthesis, while in the ß-cell it results in insulin release. It plays a crucial role in the regulation of insulin secretion and has been termed the pancreatic ß-cell sensor on account of its kinetics [4, 5]. The rate of phosphorylation of glucose in the pancreatic ß-cells is directly related to the concentration of glucose over a range of physiological glucose concentrations (4–15 mmol/l). The phosphorylation of glucose by glucokinase is the rate-limiting step in insulin secretion (Fig. 2).

Glucokinase converts glucose to glucose-6-phosphate

In the pancreatic β-cell glucokinase plays a pivotal role in regulating glucose metabolism which ultimately controls insulin secretion. Glucose-6-phosphate (end product of glucokinase activity) is channelled into the glycolytic pathway which increases the ratio of ATP/ADP. This leads to closure of the KATP channel causes insulin exocytosis
Pancreatic β-cells are able to increase their rate of glucose metabolism in response to a rise in the extracellular glucose concentration. In order for glucose to regulate its own metabolism there must be rapid equilibration across the plasma membrane and then for glucokinase to undertake glucose phosphorylation. Glucokinase has unique biochemical kinetics (demonstrates non-Michaelis-Menten kinetics) that accounts for its role as a glucose sensor [6, 7]. It has a low affinity for glucose as a substrate with a K 0.5 of ~7 mM (which is within the physiological glucose range). The enzyme is not inhibited by its end product glucose 6-phosphate and although it is a monomeric enzyme, it displays a sigmoidal saturation curve for glucose with a characteristic Hill slope of ~1.7. The inflection point of the sigmoidal curve (4−5 mM) is close to the physiological threshold for glucose-stimulated insulin secretion in β-cells. Functionally, this positive co-operativity with glucose allows the enzyme to have increased sensitivity to fluctuations in blood glucose levels. Together, these kinetic properties enable glucokinase to be highly responsive to glucose levels and to ensure that the glucose metabolic flux is closely tied to the glucose concentration.
The enzyme has at least two clefts, one for the active site, binding glucose and MgATP and the other for a putative allosteric activator [8]. Glucokinase activity is closely linked to the KATP and calcium channels of the ß-cell membrane, resulting in a threshold for glucose-stimulated insulin release of approximately 5 mmol/L, which is the set point of glucose homoeostasis.
The glucokinase gene (GCK) is located on chromosome 7p15.3–p15.1 and comprises of 12 exons which span ~45,168 bp and encode for a 465 amino acid protein with a molecular weight of 52,191 Da [9]. The gene is transcribed in various tissues but it has tissue specific promoters and is especially expressed in the pancreas, liver, and brain [10, 11]. The presence of tissue-specific promoters allows differential regulation and transcription of different transcripts giving rise to three different-sized versions of exon 1 (a, b, and c). In the pancreas the upstream promoter is functional, while in the liver the downstream promoter is used. Exon 1a is expressed in the pancreatic ß-cells whereas exons 1b and 1c are expressed in the liver.
Glucokinase gene expression in the liver is mainly regulated by insulin whereas in the ß-cell it is regulated by glucose [12, 13]. Glucokinase activity is immediately amplified by glucose metabolism in the ß-cell. Glucokinase seems to be present in two locations in the cell, cytoplasm and the granular compartment (insulin granules) with no translocation between these sites [14]. Glucokinase activity is regulated at both transcriptional and post-translational levels in ß-cells [15]. The transcription of glucokinase has been shown to be affected by a variety of factors, including insulin, cAMP, biotin, and retinoic acid [16].
The critical role of glucokinase in regulating blood glucose levels is illustrated by studies of glucokinase knockout mice and in humans with glucokinase mutations [17, 18]. Glucokinase deficient mice die shortly after birth from severe hyperglycaemia. Heterozygote (globally or in the ß-cell) glucokinase null mice survive with mild hyperglycaemia. Mice that lack glucokinase only in the liver have mild hyperglycaemia and also demonstrate defects in glycogen synthesis and glucose turnover [18]. On the other hand, over expression of GK leads to lower basal blood glucose levels and resistance to the development of high-fat-induced diabetes [19].
The unique role of glucokinase in human glucose physiology is illustrated by the fact that genetic mutations in glucokinase can either cause hyperglycaemia or hypoglycaemia (Fig. 3). Heterozygous inactivating mutations in glucokinase cause maturity-onset diabetes of the young (MODY), homozygous inactivating in glucokinase mutations result in permanent neonatal diabetes whereas heterozygous activating glucokinase mutations cause hyperinsulinaemic hypoglycaemia.

Activating glucokinase mutations increase the affinity of glucokinase for glucose leading to hypoglycaemia. In contrast inactivating glucokinase mutations reduced the affinity of glucokinase for glucose leading to hyperglycaemia
2 Glucokinase and hyperinsulinaemic hypoglycaemia
Dominant activating (or gain of function) mutations in glucokinase cause hyperinsulinaemic hypoglycaemia [7, 20–31]. Activating glucokinase mutations increase the affinity of glucokinase for glucose and alter (reset) the threshold for glucose stimulated insulin secretion [32]. Thus insulin continues to be produced at lower blood glucose levels. All reported activating mutations cluster in a region of the enzyme, which has been termed the allosteric activator site and is remote to the substrate-binding site. The allosteric site of glucokinase is where small molecule activators bind, suggesting a critical role of the allosteric site in the regulation of glucokinase activity [8]. Both glucokinase activators and activating mutations increase enzyme activity by enhancing the affinity for glucose as described by a decrease in K0.5.
The first activating missense mutation (Val455Met) in glucokinase was reported in a family with mild fasting hyperinsulinaemic hypoglycaemia [20]. Enzyme kinetics revealed that the Val455Met mutant when expressed in-vitro increased the affinity of glucokinase for glucose (by lowering of the Km for glucose by 65%). Interestingly one older family member with the mutation developed diabetes mellitus later in life, although the reasons for this are unclear. The second activating mutation (A456V) was identified in exon 10 of the glucokinase gene and mathematical modelling predicted a markedly lowered glucose stimulated insulin secretion threshold of 1.5 mmol/L [21]. There is no evidence to suggest that over-expression of glucokinase (increased gene dosage effect) is a likely cause of hyperinsulinaemic hypoglycaemia [33].
The clinical symptoms and course of patients with glucokinase mutations cover a broad spectrum from asymptomatic hypoglycaemia to unconsciousness and seizures, even within the same family with the same mutation, implicating a complex mechanism for glucokinase regulation [27, 28, 30]. Patients with activating glucokinase mutations may present with postprandial hyperinsulinaemic hypoglycaemia [27, 30]. Most of the glucokinase mutations reported to date cause mild diazoxide responsive hyperinsulinaemic hypoglycaemia. However a “de novo” mutation in glucokinase (Y214C) was described in a patient with medically unresponsive hyperinsulinaemic hypoglycaemia [23]. This mutation was located in the putative allosteric activator domain of the protein and functional studies of purified recombinant glutathionyl S transferase fusion protein of GK-Y214C showed a sixfold increase in its affinity for glucose, a lowered cooperativity, and increased kcat. The relative activity index of GKY214C was 130, and the threshold for glucose stimulated insulin secretion was predicted by mathematical modelling to be 0.8 mmol/l, as compared with 5 mmol/l in the wild-type enzyme. Other patients have now been reported with severe medically unresponsive hyperinsulinaemic hypoglycaemia due to glucokinase mutations [28]
The V91L mutation was recently reported in a patient with severe medically unresponsive hypoglycaemia [26]. The estimated threshold for glucose stimulated insulin secretion for this mutant was 0.96 mmol/L (compared to 5 mmol/L for controls). This patient required a pancreatectomy and histological examination showed that the pancreatic tissue consisted of abnormally large islets with some ß-cells containing large nuclei. Interestingly quantitative morphometric analysis indicated evidence of both proliferation and apoptosis [26]. The authors suggested that the increased glucose flux might be leading to these morphological changes.
In the largest study performed to date on a pool of patients who were negative for mutations in the ABCC8 and KCNJ11 genes the prevalence of hyperinsulinaemic hypoglycaemia due to mutations in glucokinase was estimated to be about 7% [24].
3 Glucokinase and neonatal diabetes mellitus
Homozygous mutations (inactivating) in glucokinase are a rare cause of permanent neonatal diabetes mellitus [34, 35]. Overall, inactivating glucokinase mutations have been found to reduce the phosphorylating potential of glucokinase predicting a marked decrease in both β-cell glucose usage and insulin secretion. This explains why heterozygous inactivating mutations in glucokinase account for mild fasting hyperglycaemia observed in maturity-onset diabetes of the young (MODY due to glucokinase), whereas identical mutations in the homozygous state result in the severe phenotype of permanent neonatal diabetes mellitus.
The first case of permanent neonatal diabetes mellitus due to complete glucokinase deficiency was reported in 2 patients, one of Norwegian ancestry and the other of Italian ancestry [36]. Both patients presented with intrauterine growth retardation and marked hyperglycaemia soon after birth and required subcutaneous insulin injections to control the hyperglycaemia. Investigations confirmed lack of basal insulin secretion during hyperglycaemia. Mathematical modelling predicted that in both probands with these mutations (M210K and T228M), the critical threshold for glucose stimulated insulin secretion was greater than 20 mmol/L. The parents of these subjects who were heterozygous for either the M210K or the T228M mutation had predicted thresholds for the glucose-stimulated rate of insulin secretion of about 7 mmol/L (which is characteristic of patients with glucokinase-related maturity-onset diabetes of the young MODY).
After the initial report three more patients were described by Njolstad PR et al [37]. All three patients had intrauterine growth retardation and developed insulin dependent diabetes mellitus within the first week of life. One of the patients reported was homozygous for the missense mutation Ala378Val (A378V), which is an inactivating mutation with an activity index of only 0.2% of wild-type glucokinase activity. The second patient was homozygous for a mutation in the splice donor site of exon 8 (intervening sequence 8 [IVS8] + 2T-->G), which is predicted to lead to the synthesis of an inactive protein. The third patient was a compound heterozygote with one allele having the splice-site mutation IVS8 + 2T-->G and the other the missense mutation Gly264Ser (G264S), a mutation with an activity index of 86% of normal activity.
So far a total of 10 glucokinase mutations have been reported that lead to neonatal diabetes mellitus [36–41]. All cases are homozygous or compound heterozygous for nonsense, missense or frameshift mutations and result in a deficiency of glucokinase activity. Two studies have now reported a mild beneficial effect of oral sulphonylureas in patients with glucokinase diabetes mellitus [39, 41] which seems to be dose dependent. The mechanism of sulphonylurea action in patients with homozygous GCK mutations is unclear but might involve pancreatic KATP channel dependent and independent pathways.
References
Middleton RJ. Hexokinases and glucokinases. Biochem Soc Trans. 1990;18(2):180–3.
Matschinsky FM, Magnuson MA, Zelent D, Jetton TL, Doliba N, Han Y, et al. The network of glucokinase-expressing cells in glucose homeostasis and the potential of glucokinase activators for diabetes therapy. Diabetes. 2006;55(1):1–12.
Postic C, Shiota M, Magnuson MA. Cell-specific roles of glucokinase in glucose homeostasis. Recent Prog Horm Res. 2001;56:195–217.
Matschinsky FM. Regulation of pancreatic beta-cell glucokinase: from basics to therapeutics. Diabetes. 2002;51 Suppl 3:S394–404.
Matschinsky FM, Glaser B, Magnuson MA. Pancreatic beta-cell glucokinase: closing the gap between theoretical concepts and experimental realities. Diabetes. 1998;47(3):307–15.
Iynedjian PB. Molecular physiology of mammalian glucokinase. Cell Mol Life Sci. 2009;66(1):27–42. Review.
Gloyn AL, Noordam K, Willemsen MA, Ellard S, Lam WW, Campbell IW, et al. Insights into the biochemical and genetic basis of glucokinase activation from naturally occurring hypoglycemia mutations. Diabetes. 2003;52(9):2433–40.
Grimsby J, Sarabu R, Corbett WL, Haynes NE, Bizzarro FT, Coffey JW, et al. Allosteric activators of glucokinase: potential role in diabetes therapy. Science. 2003;301:370–3.
Koranyi LI, Tanizawa Y, Welling CM, Rabin DU, Permutt MA. Human islet glucokinase gene. Isolation and sequence analysis of full-length cDNA. Diabetes. 1992;41(7):807–11.
Iynedjian PB, Möbius G, Seitz HJ, Wollheim CB, Renold AE. Tissue-specific expression of glucokinase: identification of the gene product in liver and pancreatic islets. Proc Natl Acad Sci U S A. 1986;83(7):1998–2001.
Liang Y, Jetton TL, Zimmerman EC, Najafi H, Matschinsky FM, Magnuson MA. Effects of alternate RNA splicing on glucokinase isoform activities in the pancreatic islet, liver, and pituitary. J Biol Chem. 1991;266(11):6999–7007.
Gasa R, Fabregat ME, Gomis R. The role of glucose and its metabolism in the regulation of glucokinase expression in isolated human pancreatic islets. Biochem Biophys Res Commun. 2000;268(2):491–5.
Iynedjian PB, Pilot PR, Nouspikel T, Milburn JL, Quaade C, Hughes S, et al. Differential expression and regulation of the glucokinase gene in liver and islets of Langerhans. Proc Natl Acad Sci U S A. 1989;86(20):7838–42.
Arden C, Harbottle A, Baltrusch S, Tiedge M, Agius L. Glucokinase is an integral component of the insulin granules in glucose-responsive insulin secretory cells and does not translocate during glucose stimulation. Diabetes. 2004;53(9):2346–52.
Iynedjian PB. Mammalian glucokinase and its gene. Biochem J. 1993;293:1–13.
Fernandez-Mejia C, Vega-Allende J, Rojas-Ochoa A, Rodriguez-Dorantes M, Romero-Navarro G, Matschinsky FM, et al. Cyclic adenosine 3', 5'-monophosphate increases pancreatic glucokinase activity and gene expression. Endocrinology. 2001;142(4):1448–52.
Grupe A, Hultgren B, Ryan A, Ma YH, Bauer M, Stewart TA. Transgenic knockouts reveal a critical requirement for pancreatic beta cell glucokinase in maintaining glucose homeostasis. Cell. 1995;83(1):69–78.
Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, et al. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic ß cell-specific gene knock-outs using Cre recombinase. J Biol Chem. 1999;274:305–15.
Shiota M, Postic C, Fujimoto Y, Jetton TL, Dixon K, Pan D, et al. Glucokinase gene locus transgenic mice are resistant to the development of obesity-induced type 2 diabetes. Diabetes. 2001;50:622–9.
Glaser B, Kesavan P, Heyman M, Davis E, Cuesta A, Buchs A, et al. Familial hyperinsulinism caused by an activating glucokinase mutation. N Engl J Med. 1998;338(4):226–30.
Christesen HB, Jacobsen BB, Odili S, Buettger C, Cuesta-Munoz A, Hansen T, et al. The second activating glucokinase mutation (A456V): implications for glucose homeostasis and diabetes therapy. Diabetes. 2002;51(4):1240–6.
Dullaart RP, Hoogenberg K, Rouwe CW, Stulp BK. Family with autosomal dominant hyperinsulinism associated with A456V mutation in the glucokinase gene. J Intern Med. 2004;255(1):143–5.
Cuesta-Munoz AL, Huopio H, Otonkoski T, Gomez-Zumaquero JM, Nanto-Salonen K, Rahier J, et al. Severe Persistent Hyperinsulinemic Hypoglycemia due to a De Novo Glucokinase Mutation. Diabetes. 2004;53(8):2164–8.
Christesen HB, Tribble ND, Molven A, Siddiqui J, Sandal T, Brusgaard K, et al. Activating glucokinase (GCK) mutations as a cause of medically responsive congenital hyperinsulinism: prevalence in children and characterisation of a novel GCK mutation. Eur J Endocrinol. 2008;159(1):27–34.
Barbetti F, Cobo-Vuilleumier N, Dionisi-Vici C, Toni S, Ciampalini P, Massa O, et al. Opposite clinical phenotypes of glucokinase disease: description of a novel activating mutation and contiguous inactivating mutations in human glucokinase(GCK) Gene. Mol Endocrinol. 2009;23(12):1983–9.
Kassem S, Heyman M, Glaser B, Bhandari S, Motaghedi R, Maclaren NK, et al. Large islets, beta-cell proliferation, and a glucokinase mutation. N Engl J Med. 2010;362(14):1348–50.
Meissner T, Marquard J, Cobo-Vuilleumier N, Maringa M, Rodríguez-Bada P, García-Gimeno MA, et al. Diagnostic Difficulties in Glucokinase Hyperinsulinism. Horm Metab Res. 2008 Dec 3.
Sayed S, Langdon DR, Odili S, Chen P, Buettger C, Schiffman AB, et al. Extremes of clinical and enzymatic phenotypes in children with hyperinsulinism due to glucokinase activating mutations. Diabetes. 2009;58(6):1419–27.
Pal P, Miller BG. Activating mutations in the human glucokinase gene revealed by genetic selection. Biochemistry. 2009;48(5):814–6.
Wabitsch M, Lahr G, Van de Bunt M, Marchant C, Lindner M, von Puttkamer J, et al. Heterogeneity in disease severity in a family with a novel G68V GCK activating mutation causing persistent hyperinsulinaemic hypoglycaemia of infancy. Diabet Med. 2007;24(12):1393–9. Epub 2007 Nov 1.
Christesen HB, Brusgaard K, Beck Nielsen H, Brock Jacobsen B. Non-insulinoma persistent hyperinsulinaemic hypoglycaemia caused by an activating glucokinase mutation: Hypoglycaemia unawareness and attacks. Clin Endocrinol (Oxf). 2008;68(5):747–55.
Davis EA, Cuesta-Muñoz A, Raoul M, Buettger C, Sweet I, Moates M, et al. Mutants of glucokinase cause hypoglycaemia- and hyperglycaemia syndromes and their analysis illuminates fundamental quantitative concepts of glucose homeostasis. Diabetologia. 1999;42(10):1175–86.
Van de Bunt M, Edghill ML, Hussain K, Ellard S, Gloyn A. Gene duplications resulting in over expression of glucokinase are not a common cause of hypoglycaemia of infancy in humans. Mol Genet Metab. 2008;94(2):268–9.
Vaxillaire M, Samson C, Cavé H, Metz C, Froguel P, Polak M. Glucokinase gene mutations are not a common cause of permanent neonatal diabetes in France. Diabetologia. 2002;45(3):454–5.
Gloyn AL, Ellard S, Shield JP, Temple IK, Mackay DJ, Polak M, et al. Complete glucokinase deficiency is not a common cause of permanent neonatal diabetes. Diabetologia. 2002;45(2):290.
Njølstad PR, Søvik O, Cuesta-Muñoz A, Bjørkhaug L, Massa O, Barbetti F, et al. GI. Neonatal diabetes mellitus due to complete glucokinase deficiency. N Engl J Med. 2001;344(21):1588–92.
Njølstad PR, Sagen JV, Bjørkhaug L, Odili S, Shehadeh N, Bakry D, et al. Permanent neonatal diabetes caused by glucokinase deficiency: inborn error of the glucose-insulin signaling pathway. Diabetes. 2003;52(11):2854–60.
Porter JR, Shaw NJ, Barrett TG, Hattersley AT, Ellard S, Gloyn AL. Permanent neonatal diabetes in an Asian infant. J Pediatr. 2005;146(1):131–3.
Turkkahraman D, Bircan I, Tribble ND, Akcurin S, Ellard S, Gloyn AL. Permanent Neonatal diabetes mellitus caused by a novel homozygous (T168A) glucokinase (GCK) mutation: initial response to oral sulphonylurea therapy. J Pediatr. 2008;153(1):122–6.
Rubio-Cabezas O, Diaz GF, Aragones A, Argente J, Campos-Barros A. Permanent neonatal diabetes caused by a homozygous nonsense mutation in the glucokinase gene. Pediatr Diabetes. 2008;9(3 Pt 1):245–9.
Bennett K, James C, Mutair A, Al-Shaikh H, Sinani A, Hussain K. Four novel cases of permanent neonatal diabetes mellitus caused by homozygous mutations in the glucokinase gene. Pediatr Diabetes (in press)
Author information
Authors and Affiliations
Corresponding author
Additional information
The author has nothing to declare
Rights and permissions
About this article
Cite this article
Hussain, K. Mutations in pancreatic ß-cell Glucokinase as a cause of hyperinsulinaemic hypoglycaemia and neonatal diabetes mellitus. Rev Endocr Metab Disord 11, 179–183 (2010). https://doi.org/10.1007/s11154-010-9147-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11154-010-9147-z