
Abstract
In this research, we report the preparation of doped PMo Keggin heteropolyacids where Mo is partially replaced by V, Bi, and Bi–V. These catalysts were characterized by means of ICP-AES analysis, 31P-NMR, UV–visible spectra, FT-IR spectra, thermal analysis, and textural properties. In addition, the activities of the synthesized catalysts were evaluated in the selective oxidation of sulfides to sulfoxides/sulfones. The incorporation of V, Bi and Bi–V into the structure of H3PMo12O40 increases the catalytic activity. The two most active catalysts, those with V and V–Bi were supported on aminopropyl-functionalized silica (SiO2NH2) and they were found to be and efficient heterogeneous catalysts for the selective oxidation of diphenylsulfide to the corresponding sulfoxide/sulfone.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Heteropolyacids (HPAs) are transition metal–oxygen anion clusters that exhibit a wide range of molecular sizes, compositions, and architectures [1].
There is an increasing interest in the area of heteropolycompound-induced organic transformations. In view of their remarkable catalytic properties, heteropolycompounds are applied both in bulk or supported form, and homogeneous or heterogeneous catalysis is possible.
Heteropolyacids have been used in a variety of acid-catalyzed reactions such as esterification, etherification, olefin hydration, and dehydration of alcohols, and are also attractive as catalysts for oxidation processes [2]. Recently, we have used Keggin heteropolyacids in a range of processes, i.e., preparation of heterocycles [3, 4], protection/deprotection of organic functional groups [5, 6] and oxidation processes, as well as substituted phenol, alcohol and amine oxidation, and the selective oxidation of sulfides to sulfoxides with aqueous H2O2 [7–9].
Vanadium incorporated molybdophosphoric acid (H4PMo11VO40) shows unique catalytic features for oxidation due to its bi-functional character, which arises because of the redox nature of vanadium and the oxidation/acidic character of the molybdophosphoric acid (H3PMo12O40, PMo) catalysts by replacing Mo atoms with the corresponding V atoms. These heteropolycompounds are studied due to their importance as catalysts in catalytic oxidation reactions, for example, the hydroxylation of benzene, the oxidation of toluene, nitrobenzene and norbornene using aqueous hydrogen peroxide [10], benzyl alcohol oxidation [11], benzoin oxidation to benzyls, aldehydes and esters by dioxygen [12], and liquid-phase oxidation of methane with hydrogen peroxide [13].
On the other hand, the selective oxidation of sulfides is of interest because of the importance of sulfoxides and sulfones as synthetic intermediates in organic synthesis.
Sulfoxides are important in organic synthesis as an activating group; they have been used extensively in carbon bond-forming reactions [14] as building blocks, especially as chiral auxiliaries [15], and they play key roles in enzyme activation [16]. Sulfones and their derivatives are widely applied. They can be used as intermediates for pharmaceuticals and pesticides, and for the modification of polymers [17, 18]. Certain sulfones are effective herbicides and others act as insecticides, and acaricides [19]; certain heterocyclic sulfones can be used as corrosion protection agents for metals [20].
There are several reagents available for these key transformations; they are conventionally achieved using stoichiometric amounts of both organic and inorganic reagents, for example, nitric acid, chromic acid, manganese dioxide, ozone, sodium periodate, selenium dioxide, hypervalent iodine reagents, sodium-perborate/sodium perborate [21], halogen [22], and binuclear manganese complex-periodic acid [23].
During the last decade, very useful procedures involving catalysis and hydrogen peroxide as oxidant, for example, H2WO4, H3PW12O40 + [(C8H17)4N]Br, Rhenium (V) oxo-phosphine complexes, methyltrioxorhenium, Sc (OTf)3, (salem) Mn (III), and Ti (IV) complexes, Tellurium dioxide and TPPFe(III)Cl-imidazole, have been used [24]. They have been developed to promote the oxidation of organic substrates due to their effective oxygen content, low cost, and safety in storage and operation [25, 26].
In this study, we report the preparation of doped Keggin structures where Mo is partially replaced by V, Bi, and Bi–V, respectively, in the primary structure of PMo. The activities of the synthesized catalysts were evaluated in the selective oxidation of sulfides to sulfoxides, using aqueous hydrogen peroxide as oxidant. Two bulk HPAs (those doped with V and VBi) were supported on aminopropyl-functionalized silica (SiO2NH2) and they were prepared by the equilibrium adsorption technique. These catalysts were well characterized by means of 31P-NMR, UV–visible spectra, FT-IR spectra, thermal analysis, and textural properties. Mo, V and Bi amounts were estimated by ICP-AES analysis.
Experimental
Catalyst synthesis
Heteropolyacid synthesis. Bulk heteropolyacid synthesis
The catalysts were prepared by the hydrothermal synthesis method [27]. The following HPA synthesis procedure was used: a stoichiometric mixture of MoO3, the corresponding metal oxide and H3PO4 [85% (w/v)] were suspended in 120 mL of distilled water. The mixture was stirred for 3 h at 75 °C. After cooling down to room temperature and removal of insolubles, the heteropolyacid solution was evaporated and dried at 40 °C. Colorful crystals of pure catalysts were obtained. The used nomenclature was: -doped with V: PMoV, -doped with Bi: PMoBi, and -doped with V–Bi: PMoVBi.
The experimental contents of Mo, V and Bi in the bulk HPA were determined by means of the inductively coupled plasma atomic emission spectroscopy (ICP-AES) technique using a Shimadzu 1000 III instrument.
Supported PMoV, and PMoBiV synthesis. Silica preparation via sol–gel technique
Tetraethoxysilane (TEOS) (102 mL), absolute ethanol (99.99%) (42 mL), and glacial acetic acid (30 mL), in a molar ratio equal to 1:1.6:1.1, were mixed in a glove box under nitrogen atmosphere at room temperature. The mixture was removed from the controlled atmosphere and another part of absolute ethanol (90 mL) was added, and then sol gelation took place. Finally, wet gel was aged in the same medium until dry silica particles were obtained. These particles were dried at room temperature and this solid was named SiO2 [specific surface area (S BET): 292.1 m2/g].
Preparation of functionalized silica
SiO2 (4 g) was refluxed with toluene (128 mL) for 1 h. Then, 3-aminopropyltrimethoxysilane (APS) (6.40 mL) was added and stirred under reflux conditions for 24 h. The solid was filtered, washed in a Soxhlet apparatus with diethyl ether and dichloromethane and dried at 40 °C according to Ref. [28]. These particles were dried at 40 °C and this solid was named SiO2NH2 [the specific surface area (S BET) near of about 2 m2/g, this value is low and near to experimental error], and the –NH2 content was estimated to be near 3.5 mmol per g of SiO2.
Preparation of silica-supported catalysts
Supported catalysts were obtained by the equilibrium adsorption technique. The solutions for impregnation were prepared by dissolution of the HPAs (PMoV and PMoVBi, 160 mg of each one) in absolute ethanol (3.50 mL). The first step in the impregnation technique was to contact the HPA solution with the support SiO2NH2 (500 mg), with addition of H2O (0.50 mL). The support with impregnated solution was left in contact during 24 h.
The two new catalysts were named SiO2NH2PMoV and SiO2NH2PMoVBi. The final contents of HPAs in the catalysts SiO2NH2PMoV and SiO2NH2PMoVBi were 24.5 and 23.8% (w/w), respectively.
Catalyst characterization
Inductively coupled plasma atomic emission spectroscopy (ICP-AES)
The experimental contents of Mo, V and Bi in the bulk HPA were determined by means of the inductively coupled plasma atomic emission spectroscopy (ICP-AES) technique using a Shimadzu 1000 III instrument.
31P nuclear magnetic resonance (31P-NMR)
Solid samples were analyzed by 31P MAS NMR by means of Varian Mercury Plus 300 equipment with a sample holder 7 mm in diameter, a resonance frequency of 121.469 MHz, and a spinning rate 5 kHz. The measurements were carried out at room temperature using 85% (w/v) H3PO4 as external reference.
UV–visible spectroscopy (UV–VIS)
UV–visible spectra of solutions in quartz cuvettes were measured at room temperature with a Perkin Elmer Lambda 35 UV–vis double beam spectrophotometer, in the range 200–1,100 nm. This spectrometer uses tungsten and deuterium lamps to provide visible and UV wavelengths. Ethanolic solutions of 0.001 M concentration were chosen, and all samples were soluble at this concentration. The adsorption edge for each compound was defined by extrapolating the region of steep descent in the spectrum to zero, as shown in Fig. 1 for H3PMo12O40, using the method reported by Barteau et al. [29].

UV–visible spectrum of 0.001 M ethanolic solution of PMo
Fourier transform infrared spectroscopy (FT-IR)
Thermo Nicolet IR.200 equipment, pellets in BrK, and a measuring range of 400–4,000 cm−1 were used to obtain the FT-IR spectra of the solid samples at room temperature.
Textural properties
Specific surface area (S BET): the pore volume and the mean pore diameter of the supported catalysts were determined by nitrogen adsorption/desorption techniques using Micromeritics ASAP 2020 equipment at liquid-nitrogen temperature. The sample was previously degassed at 100 °C for 1 h.
Catalytic activity test
General
All reagents were purchased from Merck and Aldrich and used without further purification. All yields refer to isolated products after purification. The products were characterized by spectroscopy data (1H and 13C-NMR). The NMR spectra were recorded on a 200 MHz Bruker spectrometer. The NMR spectra were measured in CDCl3 relative to TMS (0.00 ppm). The organic phase was dried on anhydrous Na2SO4 and filtered for its analysis by gas chromatography using Varian Scan 3400 cx equipment. The product distribution was quantified by a Shimadzu C-R34 instrument. Reactions were monitored by thin layer chromatography (TLC) analyses.
Diphenylsulfide oxidation
Under homogeneous conditions
Selective oxidation of diphenylsulfide to diphenylsulfoxide Bulk HPA (6 mg, ~0.003 mmol), diphenylsulfide (0.5 mmol, 93 mg), and ethanol (2 mL) were stirred at 25 °C. Then aqueous H2O2, 35% (w/v) (0.65 mmol, 0.075 mL), was added. The progress of the reaction was monitored by TLC and GC–MS analysis. After the end of the reaction, it was treated with dichloromethane (10 mL) and water (2 × 3 mL). The organic phase was dried on anhydrous Na2SO4 and concentrated to obtain diphenylsulfoxide. The molar ratio between diphenylsulfide and H2O2 35% (w/v) was 1:1.3.
Selective oxidation of diphenylsulfide to diphenylsulfone Diphenylsulfone was obtained using bulk HPAs (12 mg, ~0.006 mmol), diphenylsulfide (1 mmol, 186 mg), 35% (w/v) H2O2 (6.60 mmol, 0.70 mL), and ethanol (4 mL) at 50 °C.
The molar ratio between diphenylsulfide and H2O2 35% (w/v) was 1:6.6.
Under heterogeneous conditions
Selective oxidation of diphenylsulfide to diphenylsulfoxide The supported catalyst (25 mg), diphenylsulfide (0.5 mmol, 93 mg), and ethanol (2 mL) were stirred at 25 °C. Then 35% (w/v) H2O2 (0.65 mmol, 0.075 mL) was added. The progress of the reaction was monitored by TLC and GC–MS analysis. After the end of the reaction, the catalyst was recovered by centrifugation and washed with dichloromethane (10 mL). The reaction mixture was treated with dichloromethane (10 mL) and water (2 × 3 mL). The organic phase was dried on anhydrous Na2SO4 and concentrated to obtain diphenylsulfoxide.
The molar ratio between diphenylsulfide and H2O2 35% (w/v) was 1:1.3.
Selective oxidation of diphenylsulfide to diphenylsulfoxide or diphenylsulfone Diphenylsulfone was obtained similarly using 0.375 mL of H2O2 35% (w/v) at 50 °C.
The molar ratio between diphenylsulfide and H2O2 35% (w/v) was 1:6.5.
Recycling of catalyst After reaction, the catalyst was filtered, washed with ethanol, dried under vacuum, and reused for the next cycle of oxidation reaction, following the procedure described above.
Results and discussion
Catalysis characterization
The experimental contents of Mo, V and Bi in the bulk HPA were determined by means of the inductively coupled plasma atomic emission spectroscopy (ICP-AES) technique using a Shimadzu 1000 III instrument. The analysis of the prepared heteropolyacids is as follows:
H4PMo11VO40·12H2O. Calculated (theoretical): Mo, 52.83, V, 2.55. Found: Mo, 52.85, V 3.4.
H6PMo11O40BiO40·12H2O. Calculated (theoretical): Mo, 46.96, Bi, 9.30. Found: Mo, 47.45, Bi, 9.36.
H5PMo11V0.5Bi0.5·12H2O. Calculated (theoretical): Mo, 50.92, V 1.23, Bi, 5.04. Found: Mo, 50.55, V 3.51, Bi 0.16.
The experimental results show some deviations of the calculated values. This is due to possible formation of a mixture of compounds during the one-pot synthesis. For example, PMoV contains a higher amount of vanadium than the expected, which indicates the presence as the admixture of H5PV2M10O40. Similarly, for the PMoVBi catalyst, the elemental composition corresponds to the probable formation of H4PVMo11O40 with a little higher admixture of H5PV2Mo10O40 than in the first case, and of a small amount of a separate PMoBi compound.
The results obtained by 31P-MAS-NMR are listed in Table 1. All the chemical shifts appear at higher magnetic fields than that of the external reference used (H3PO4). The spectrum of bulk PMo has one sharp line at −3.61 ppm. A similar result was obtained for PMoBi (−3.64 ppm) and PMoV and PMoBiV have one sharp line at −3.20 and 3.17, respectively [30].
Fig. 1 shows UV–visible spectra of the bulk PMo solutions. The band attributed to oxygen–metal transfers at 210–260 nm can be seen in all the spectra. Moreover, Table 1 includes all the absorption edges of HPAs measured in this study. The introduction of V into the framework of Keggin ion heteropolyacids results in lower absorption edge energies in UV–visible spectra. This property is conserved when Bi and V are included during HPA synthesis. But when Bi is only incorporated in the synthesis, the absorption edge energy is near that of PMo.
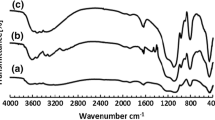
The results obtained by FT-IR spectra of bulk HPAs are shown in Fig. 2. The vibration spectra of the bulk HPAs with Keggin structure are modified as a function of the nature of the elements introduced into their structure. The FT-IR spectrum of PMo has been previously studied [31], the main bands were observed at 1,064 (P–Oa), 962 (Mo–Od), 871 (Mo–Ob–Mo), and 780 (Mo–Oc–Mo) cm−1. In the FT-spectrum of PMoV, bands at 1,061 with a shoulder at 1,081 (P–Oa), 960 (Mo–Od), 863 (Mo–Ob–Mo), and 777 (Mo–Oc–Mo) cm−1 were observed, as reported by Villabrille and coworkers [30]. In the FT-IR spectrum of PMoBi the main bands were observed at 1,063 (P–Oa), 966 (Mo–Od), 872 (Mo–Ob–Mo), and 789 (Mo–Oc–Mo) cm−1, and in PMoBiV the principal bands were observed at 1,061 (P–Oa), 962 (Mo–Od), 865 (Mo–Ob–Mo), and 780 (Mo–Oc–Mo) cm−1. When another atom is introduced into the PMo structure, the same characteristic bands are present in the spectra, which confirm the Keggin structure, but the difference in length of the bonds could be introducing defects into the original spectrum. Here it is important to consider the atomic properties of V and Bi. Although the small displacements that take place in some bands (Fig. 2), as shown for Mo–Ob–Mo, could be due to many different reasons during the incorporation of V, Bi or V–Bi into the structure, they are usually attributed to vibrations of the bridges between “inter” (Mo–Ob–Mo) and “intra” (Mo–Oc–Mo) groups (Mo3O13).

FT-IR spectra of bulk PMo, PMoV, PMoBiV and PMoBi catalysts
For a better definition of the species resulting from the interaction of both heteropolyacids with the impregnated support in equilibrium, they were characterized by FT-IR. In Fig. 3, FT-IR spectra of the functionalized support (SiO2NH2), bulk PMoV and PMoBiV, and supported catalysts (SiO2NH2PMoV) and (SiO2NH2PMoBiV) are shown. The characteristic spectrum of silica shows bands at 1,000, 800, and 470 cm−1; there are other bands at 3,400 and 1,620 cm−1 attributed to the stretching and bending of the OH groups, respectively [32]. In relation to functionalized silica, the main differences are the N–H stretching (band 3,300–2,600 cm−1) broad peak overlapped with the silanol stretching bands of silica and –CH2– groups. The small shoulders at the 3,000–2,750 cm−1 region can be assigned to C–H stretching of the amino propyl group at the silica surface. Also, the functionalizing agent (APS) used in the grafting process presents a strong band at the 1,110–1,050 cm−1 region attributed to Si–O–C aliphatic groups, which is observed as a transmittance increase of the silica band in the functionalized support. In addition, the APS band at 790 cm−1 is present without overlapping and a transmittance increase at 550 cm−1 can be seen on the silica band [33].

FT-IR spectra of supported SiO2–NH2–PMoV and SiO2–NH2–PMoBiV catalysts
During the equilibrium impregnation technique, there is an electrostatic interaction between amino groups and bulk HPAs. By this characterization technique it is very difficult to obtain a band corresponding to PMoV due to overlapping with other bands of the support.
N2 adsorption tests were used to give a textural characterization of the catalysts and the results [S BET (m2/g)] are listed in Table 2. The S BET areas for bulk HPAs are low, between 2.7 and 13.8 m2/g (Table 2, entry 1–4). Supported catalysts SiO2NH2PMoV and SiO2NH2PMoVBi show a slightly lower surface area (4.4 m2/g). This effect can be attributed to an electrostatic interaction between NH2 groups and HPA proton, as mentioned in the previous paragraph, due to an umbrella effect of APS [33] on silica, and then the specific surface area is reduced for bulk HPA impregnation.
Catalytic test
Selective oxidation of diphenylsulfide
Initially, the catalytic performances of the different bulk synthesized heteropolyacids (PMoV, PMoBi, and PMoVBi) were evaluated in a homogeneous oxidation reaction. The oxidation of diphenylsulfide with aqueous hydrogen peroxide [35% (w/v)] was selected as the model reaction (Scheme 1). Ethanol was chosen as the solvent of the reaction because it gives a homogeneous phase with the reagents and it is a nontoxic and safe solvent.

Selective oxidation of sulfide to sulfoxide and/or sulfone
We first screened the selective oxidation of diphenylsulfide to diphenylsulfoxide using the different catalysts. The blank experiment was performed in the absence of the catalyst using an almost stoichiometric amount of aqueous H2O2 (35% (w/v), 0.075 mL, 0.78 mmol). Under these conditions, the reaction conversion was very low (5% for 7 h), at 25 °C (Fig. 4), but when an HPA is added, the conversion increases to values between 58 and 75%.

Catalytic performance of different HPAs in diphenylsulfide oxidation. Sulfoxide and sulfone selectivity (%) versus sulfide conversion (%). Experimental conditions: bulk HPA (6 mg), 0.7 mmol of diphenylsulfide, H2O2, 35% (w/v) (0.65 mmol, 0.075 mL), and 2 mL of ethanol, reaction for 7 h at 25 °C
For these conditions, PMoV and PMoBiV showed the best performance. When PMoV was used, a conversion of 74% was observed at 7 h, with 94% of sulfoxide selectivity. Similarly, PMoVBi showed a conversion and selectivity of 75 and 90%, respectively, in this time period. Under the same reaction conditions, PMo only gives a sulfide conversion of 58%. In all experiments performed, the synthesized catalyst showed more activity and selectivity in the diphenylsulfide oxidation to sulfoxide than the corresponding PMo, especially when vanadium was incorporated. The PMoV catalyst shows unique catalyst features for oxidation due to its bifunctional character, which arises because of the redox nature of Vanadium and the oxidation/acidic character of the molybdophosphoric acid [34]. As reported by Song et al. [1], the metal substitution may be modifying the energy and composition of the lowest unoccupied molecular orbital (LUMO) and consequently its redox properties. The substitution of vanadium ions into the molybdenum framework stabilizes the LUMOs because these orbitals derive in part from vanadium d-orbitals, which have been assumed to be more stable than those of Molibdenum and Tungsten. In this direction, Barteau et al. [29] reported that the absorption edge in the UV–visible spectrum of HPAs measures the energy required for ligand-to-metal charge transfer (LMCT). This represents the transfer of an electron from the highest occupied molecular orbital (HOMO) to the LUMO. Because the HOMO involves mostly the terminal oxygens, its energy is not greatly affected by changes in the HPA framework. However, the LUMO is greatly affected because it involves the bridging oxygen and the d-orbitals of the framework metals. Thus, changes in the absorption edge largely reflect changes in the LUMO [29]. The use of UV–visible spectroscopy of HPAs in solution could be a simple diagnosis of the redox properties.
Fig. 5 shows the correlation between the conversion (%) of diphenylsulfide and the adsorption edge of the HPA catalyst in solution. HPAs having adsorption edge energies below 2.31 eV displayed significant catalytic activity. As adsorption edge energies dropped from 2.31 to 2.26 eV with framework vanadium substitution into these Keggin HPAs, there was a general increase in sulfide conversion (Fig. 5).

Sulfide conversion (%) in the selective oxidation of diphenylsulfide to diphenylsulfoxide versus absorption edge energies for HPA ethanol solution. Experimental conditions: bulk HPA (6 mg), 0.7 mmol of diphenylsulfide, H2O2, 35% (w/v) (0.65 mmol, 0.075 mL), and 2 mL of ethanol, reaction for 7 h at 25 °C
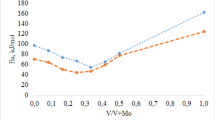
Then, we performed the selective oxidation of diphenylsulfide to diphenylsulfone using an excess of H2O2 (35% (w/v), 0.7 mL, 6.6 mmol) at 50 °C. Under these conditions, it is possible to obtain diphenylsulfone selectively. All the doped catalysts presented a similar catalytic activity, which was higher than that of PMo. At short reaction times, when the PMoBi catalyst is used (Fig. 6), a 24% of sulfone selectivity is observed at 1 h, with a 100% of sulfide conversion. The sulfone selectivity for PMoV and PMoVBi catalysts is similar (30 and 35%, respectively), and for PMo it is only 12% under the same reaction conditions (Fig. 6). There is also a good correlation between sulfone selectivity (%) and the adsorption edge of the HPA catalyst in solution (Fig. 7).

Catalytic performance of different HPAs in diphenylsulfide oxidation. Sulfone selectivity (%). Experimental conditions: bulk HPA (12 mg), 1 mmol of diphenylsulfide, H2O2, 35% (w/v) (6.6 mmol, 0.7 mL), and 4 mL of ethanol, reaction for 1 h at 50 °C. Sulfide conversion 1 h

Sulfone selectivity (%) in the oxidation of diphenylsulfide to diphenylsulfone versus absorption edge energies for HPA ethanol solution. Experimental conditions: bulk HPA (12 mg), 1 mmol of diphenylsulfide, H2O2, 35% (w/v) (6.6 mmol, 0.7 mL), and 4 mL of ethanol, reaction for 1 h at 50 °C. Sulfide conversion 1 h
Although HPA catalysts show a good performance in this reaction, the bulk HPAs are completely soluble in the reaction medium, and their isolation and reuse are difficult. In order to carry out the reaction under heterogeneous conditions, the more active HPAs were supported on amino-functionalized silica. Fig. 8 shows the results obtained using PMoV and PMoVBi supported on silica (SiO2NH2PMoV and SiO2NH2PMoVBi), in the selective diphenylsulfide oxidation to diphenylsulfoxide at 25 °C. Silica-supported PMo was studied previously in the selective oxidation of sulfide to sulfones [35]. The PMoVBi catalyst supported on silica (SiO2NH2PMoVBi) shows the best performance, sulfide conversion is low and sulfoxide selectivity is good. When this catalyst is used under the selective diphenylsulfide oxidation to sulfoxide conditions, a conversion of only 28% is observed at 27 h, with a 55% of sulfoxide selectivity (Fig. 8). The catalytic activity of selected HPAs was tested at 50 °C using excess of H2O2 with the objective of obtaining diphenylsulfone selectivity. Both materials (SiO2NH2PMoV and SiO2NH2PMoVBi) are excellent catalysts in the selective oxidation of diphenylsulfide to the corresponding sulfoxide or sulfone, depending on the reaction time. Fig. 9 shows the diphenylsulfide conversion versus the reaction time. The SiO2NH2PMoV and SiO2NH2PMoVBi catalysts give a sulfide conversion and selectivity of 100% in a reaction time of 2 h and 4 h, respectively.

Catalytic performance of SiO2NH2PMoBiV and SiO2NH2PMoV in the selective oxidation of diphenylsulfide. Sulfide conversion (%) versus time (h). Experimental conditions: the supported HPA (25 mg), 0.5 (93 mg) mmol of diphenylsulfide, H2O2, 35% (w/v) (0.65 mmol, 0.075 mL), and 2 mL of ethanol, reaction for 26 h at 25 °C

Catalytic performance of SiO2NH2PMoBiV and SiO2NH2PMoV in the selective oxidation of diphenylsulfide. Sulfide conversion (%) versus time (h). Experimental conditions: the supported HPA (25 mg), 0.5 mmol of diphenylsulfide (93 mg), H2O2, 35% (w/v) (6.6 mmol, 0.375 mL), and 2 mL of ethanol, reaction for 8 h at 50 °C
In addition, one of the main advantages of using supported solid catalyst in the liquid-phase reaction is the ease of separation and reuse of the catalyst in the catalytic cycles. In this way, the catalyst (SiO2NH2PMoV) was separated by filtration, washed with ethanol, dried and then reused. No significant changes were observed after three catalytic cycles.
Conclusions
In this research, we report the preparation of doped Keggin structures with V, Bi and Bi–V, where Mo is partially replaced by those elements in molybdophosphoric acid (PMo). These catalysts were well characterized by means of FT-IR, 31P-NMR, and UV–visible spectra. Mo, V and Bi amounts were estimated by ICP-AES analysis. In addition, the activities of the synthesized catalysts were evaluated in the selective oxidation of sulfides to sulfoxides/sulfones.
The correlation between catalytic oxidation performance (sulfide oxidation) and absorption edge energies of HPA catalysts demonstrated that the absorption edge energies could be utilized as a correlation parameter for the reduction potentials (oxidizing powers) of the HPA catalysts, and furthermore, as a probe of catalytic oxidation performance of the HPA catalysts.
Then, the two most active catalysts were immobilized on aminopropyl-functionalized silica. The materials prepared by the equilibrium adsorption technique (SiO2NH2PMoV and SiO2NH2PMoVBi) were found to be efficient, ecofriendly and recyclable heterogeneous catalysts for the selective oxidation of diphenylsulfide to the corresponding sulfoxide/sulfone, under mild reaction conditions using aqueous hydrogen peroxide as the oxidant. The incorporation of V, Bi and Bi–V into the structure of PMo increases the catalytic activity.
References
Park D, Park P, Bang Y, Song I (2010) Appl Catal A 373:201–207
Romanelli G, Autino J (2009) Mini Rev Org Chem 6:359–366
Bennardi D, Romanelli G, Autino J, Pizzio L, Vázquez P, Cáceres C, Blanco M (2010) React Kinet Mech Catal 100:165–174
Romanelli G, Autino P, Vázquez P, Pizzio L, Blanco M, Cáceres C (2003) Appl Catal A 352:208–213
Villabrille P, Romanelli G, Quaranta N, Vázquez P (2010) Appl Catal B 96:379–386
Sambeth J, Romanelli G, Autino J, Thomas H, Baronetti G (2010) Appl Catal A 378:114–118
Tundo P, Romanelli G, Vázquez P, Aricó F (2010) Catal Commun 11:1181–1184
Tundo P, Romanelli G, Vázquez P, Loris A, Aricó F (2008) Synlett 7:967–970
Villabrille P, Romanelli G, Vázquez P, Cáceres C (2008) Appl Catal A 334:374–380
Rao P, Venkateswara Rao K, Sai Prasad P, Lingaiah N (2010) Catal Commun 11:547–550
Kende A, Ebertino F (1984) Tetrahedron Lett 25:923–926
El Ali B, El-Ghanam A, Fettouhi M (2001) J Mol Catal A 165:283–289
Seki Y, Mizuno K, Misono M (2000) Appl Catal A 194:13–20
Trost B (1978) Chem Rev 78:363–382
Carreno M (1995) Chem Rev 95:1717–1760
Karami B, Ghoreishi-Nezhad M, Clark J (2005) Org Lett 8:625–628
Ogura K, Yahata N, Fujimori T, Fujita M (1990) Tetrahedron Lett 31:4621–4624
Ma Y, Liu R, Gong X, Li Z, Huang Q, Wang H, Song G (2006) J Agric Food Chem 54:7724–7728
Friedrich M, Meichle W, Bernhard H, Rihs G, Otto H (1996) Archiv der Pharmazie 329:361–370
Barrett A, Carr R, Attwood S, Richardson G, Walshe N (1986) J Org Chem 51:4840–4856
Varma R, Sain R, Meshram H (1997) Tetrahedron Lett 8:625–628 and references cited herein
Tajbakhsh M, Hosseinzadeh R, Shakoori A (2004) Tetrahedron Lett 45:1889–1893
Barton D, Li W (1998) Tetrahedron Lett 39:7075–7078
Iwahana T, Sakaguchi S, Ishii Y (1998) Tetrahedron Lett 39:9059–9062
Bonadies F, De Angelis F, Locati L, Scettri A (1996) Tetrahedron Lett 37:7129–7130
Kaczorowska K, Kolarska Z, Mitka K, Kowalski P (2005) Tetrahedron 61:8315–8327
Villabrille P, Romanelli G, Vázquez P, Cáceres C (2004) Appl Catal A 270:101–111
Tarlani A, Abedini M, Nemati A, Khabaz M, Amini M (2006) J Colloid Interface Sci 303:32–38
Barteau K, Lyons J, Song I, Barteau M (2006) Top Catal 41:55–62
Villabrille P, Romanelli G, Gassa L, Vázquez P, Cáceres C (2007) Appl Catal A 324:69–76
Vázquez P, Blanco M, Cáceres C (1999) Catal Lett 60:205–215
Vázquez P, Pizzio L, Cáceres C, Blanco M, Thomas H, Alesso E, Finkielsztein L, Lantaño B, Moltrasio G, Aguirre J (2000) J Mol Catal A 161:223–232
Pizzio L, Romanelli G, Vázquez P, Autino J, Blanco M, Cáceres C (2006) Appl Catal A 308:153–160
Casarini D, Centi G, Jiro P, Lena V, Tvaruzkova Z (1993) J Catal 143:325–344
Palermo V, Vázquez P, Romanelli G (2009) Phosphorus Sulfur Silicon Relat Elem 12:3258–3268
Acknowledgments
The authors thank E. Soto and D. Peña for their experimental contribution to the measurements of S BET and GC, respectively. The authors thank CONICET, ANPCyT and UNLP for their financial support. AGS, PGV, HJT and GPR are members of CONICET.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Palermo, V., Sathicq, Á.G., Vázquez, P.G. et al. Doped Keggin heteropolyacids as catalysts in sulfide oxidation. Reac Kinet Mech Cat 104, 181–195 (2011). https://doi.org/10.1007/s11144-011-0341-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-011-0341-0